par Patrick PLA, Université Paris Saclay
- La culture cellulaire
- Fractionnement cellulaire par centrifugation différentielle
- Western-blot
- Immunoprécipitation
- Le double-hybride
- Immunohistochimie/Immunocytochimie/Immunofluorescence
- Les progrès de la microscopie et de l’analyse des images
- Approches optogénétiques pour perturber la signalisation cellulaire
- Les rapporteurs fluorescents pour la concentration calcique
- La cytométrie de flux
- La microscopie électronique
- Les tests de migration cellulaire
- Les marqueurs de prolifération
- L’étude du cycle cellulaire
La culture cellulaire
La culture cellulaire in vitro concerne :
- des cultures primaires : c’est-à-dire des cellules prélevées sur un être vivant et que l’on met en culture in vitro mais qui ne peuvent pas être maintenues sur une longue durée de temps avec une prolifération élevée (entre autre, parce qu’elles atteignent la limite de Hayflick où elles entrent en sénescence, notamment à cause du raccourcissement des télomères à chaque réplication).
- des lignées cellulaires : c’est-à-dire des cellules prélevées sur un être vivant il y a longtemps et qui maintiennent des capacités de prolifération théoriquement illimitées (parce qu’elles sont cancéreuses, ou parce qu’elles ont des propriétés de cellules souches ou parce qu’elles ont été « immortalisées » en leur faisant exprimer un oncogène (souvent l’antigène T du virus SV40 qui inhibe Rb et p53).
Les cellules HeLa, isolées en 1951 à partir d’un carcinome du col de l’utérus d’Henrietta Lacks (sans son consentement), ont constitué la première lignée cellulaire humaine immortelle que les chercheurs ont pu faire se diviser indéfiniment in vitro. Malgré les problèmes éthiques liés à leur prélèvement, elles demeurent un outil fondamental en biologie cellulaire, apparaissant dans plus de 120 000 publications scientifiques. D’autres lignées cellulaires cancéreuses ont depuis été établies, cette fois-ci avec le consentement des donneurs.
Des stocks de lignées cellulaires peuvent être conservées « indéfiniment » dans de l’azote liquide (cryoconservation à -196°C).
La culture cellulaire présente de nombreux avantages mais aussi des inconvénients. Les cellules sont dans un environnement contrôlé (température, nutriments disponibles, éventuellement en présence de matrice extracellulaire…) mais cet environnement est parfois éloigné des conditions physiologiques. Dans des cultures classiques en 2D chaque cellule est facilement visible, mais les propriétés des organisations 3D dans les tissus peuvent être perdues. Les capacités importantes de prolifération, notamment des lignées cellulaires utilisées permettent d’obtenir beaucoup de matériel pour faire des études de biochimie et facilitent les analyses statistiques mais les cellules ne sont pas dans leur cadre naturel et des cellules très proliférantes ne correspondent qu’à un profil particulier de cellules.
La culture cellulaire permet de diminuer le recours à l’expérimentation animale mais ne peut pas la remplacer complètement.
Les cellules sont cultivées sur du plastique traité pour que les cellules puissent y adhérer ou sur des éléments de matrice extracellulaire que l’on dépose avant d’ensemencer les cellules (collagène, fibronectine ou encore le Matrigel (lame basale produite par des cellules de sarcome de souris) qui par son épaisseur permet de créer un environnement 3D).
Les cellules sont cultivées dans un milieu nutritif contenant des nutriments et des vitamines et souvent additionné de sérum (en général du sérum de veau foetal) dans lequel se trouvent des facteurs de croissance nécessaires à la prolifération. Pour les cellules qui prolifèrent, on suit le niveau de confluence, c’est-à-dire le pourcentage de la surface couvert par les cellules.
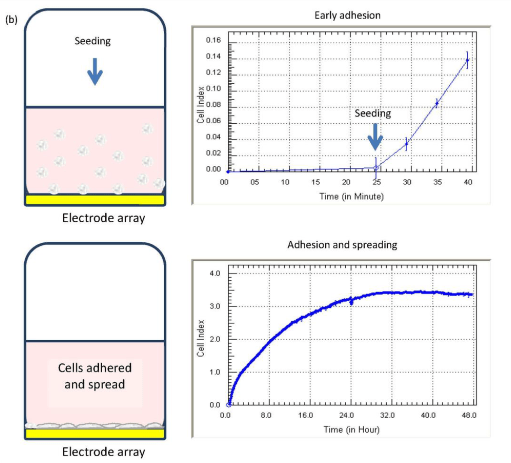
Des nouvelles technologies permettent un suivi en continu de l’adhérence, de la prolifération et de la migration des cellules lors de la culture.
Le passage des cellules confluentes en culture cellulaire consiste à détacher, à diluer puis à redéposer des cellules dont la densité avait atteint le maximum autorisé sur la surface de culture. Lorsque les cellules deviennent confluentes, leur prolifération ralentit en raison du contact cellulaire et du manque d’espace. Pour maintenir leur état physiologique et leur capacité de division, on procède à une dissociation mécanique et/ou enzymatique (souvent à la trypsine-EDTA (la trypsine digère la matrice extracellulaire et l’EDTA piège les ions Ca2+ nécessaires aux cadhérines qui permettent l’adhérence des cellules entre elles), suivie d’un comptage et d’un repiquage dans de nouveaux flacons à une densité adaptée. Cette étape est essentielle pour préserver la viabilité et la stabilité des lignées cellulaires au fil du temps.
Fractionnement cellulaire par centrifugation différentielle
Il s’agit d’une technique pour purifier des fractions particulières du contenu des cellules (noyau, mitochondries, ribosomes…). Elle est basée sur une série de centrifugation à des vitesses de plus en plus élevées qui permettent de précipiter des composants de plus en plus petits.
Exemple d’application du fractionnement cellulaire :
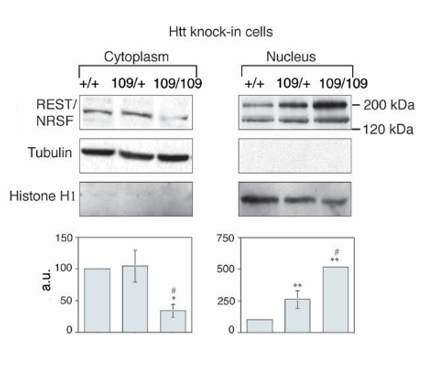
Western-blot
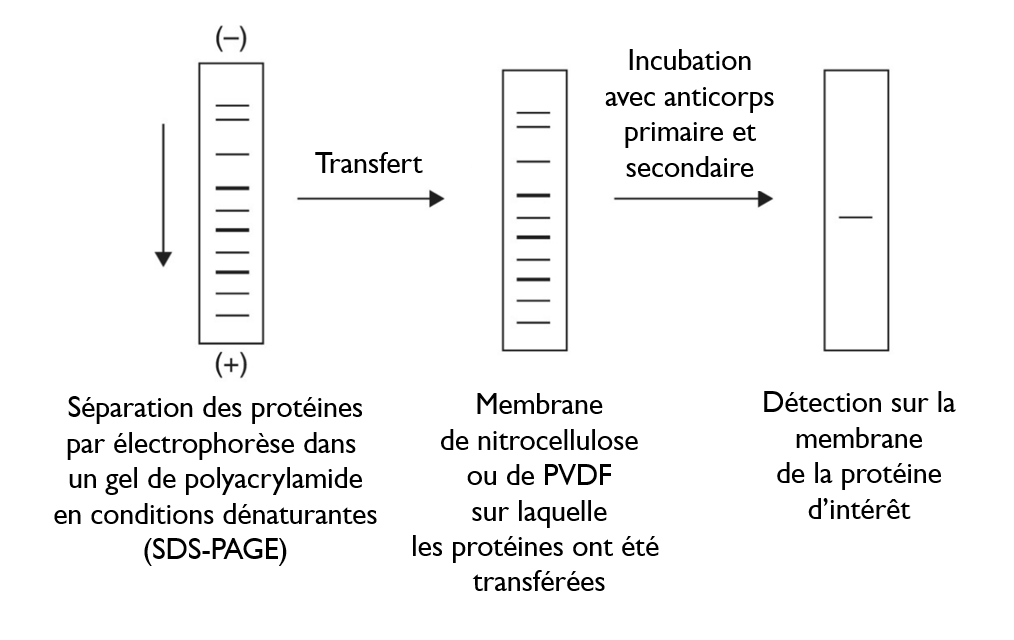
Il s’agit d’une méthode de séparation des protéines selon leur taille dans un gel de polyacrylamide puis de transfert vers une membrane où les protéines sont identifiées grâce à des anticorps.
Les anticorps utilisés (pour le western-blot ou d’autres techniques utilisant des anticorps) peuvent être de deux types :
- Des anticorps polyclonaux (en fait un mélange d’anticorps différents) qui sont produits par un animal (lapin, souris, chèvre…) après injection de l’antigène que l’on souhaite reconnaître.
- Des anticorps monoclonaux (tous identiques) contre un antigène donné produits dans des hybridomes, c’est-à-dire des lymphocytes B rendus « immortels » après fusion avec des cellules cancéreuses de myélome.
Présentation de la technique de western-blot en vidéo :
Un exemple de protocole d’extraction de protéines et de western-Blot en détail :
Les cellules ont été lysées dans du tampon RIPA (150 mM NaCl, 1 % NP-40, 0,25 % Na-désoxycholate, 50 mM Tris-HCl [pH 7,4]) contenant 1 comprimé
d’inhibiteur de protéase et 1 comprimé d’inhibiteur de phosphatase par 10 ml. La concentration totale de protéines dans les lysats a été déterminée à l’aide d’un kit Pierce BCA Protein Assay. L’absorbance a été mesurée à l’aide d’un spectrophotomètre à microplaque. Un gel de polyacrylamide à 8 % contenant du SDS surmonté d’un gel de stacking (pour concentrer les protéines) a été préparé. Ensuite, 15 µL d’échantillon, contenant 9 µg de protéines totales et 5 µL de tampon d’échantillon (5,7 ml d’eau, 1,6 ml de glycérol (densité importante pour que l’échantillon aille au fond du puits), 1,1 ml de SDS à 10 % (dénaturation), 1,3 ml de Tris 0,5 M (pH 6,8), 25 mg de dithiotréitol (DTT, casse les ponts disulfures des protéines), 300 µL de bleu de bromophénol (pour bien visualiser l’échantillon)) ont été chauffés à 90 °C pendant 5 min (pour dénaturer les protéines), refroidis sur de la glace et chargé sur le gel. Le gel a été incubé dans du tampon d’électrophorèse (Tris Base 25 mM, glycine 190 mM, 0,1 % SDS) à 70 V jusqu’à ce que les échantillons atteignent le gel de séparation, puis à 150 V jusqu’à ce que les échantillons atteignent le bas du gel. Ensuite, les protéines ont été transférées sur une membrane en polyfluorure de vinylidène (PVDF) pendant 1 h à 80 V sur glace dans un tampon de transfert froid (Tris base 25 mM, glycine 190 mM, éthanol 20%). La membrane a été rincée à l’eau et lavée 2x dans une solution saline tamponnée au Tris et du Tween-20 (TBS-T) (20 mM de Tris/HCl, 137 mM de NaCl, 0,1 % de Tween-20). La membrane a été incubée pendant 1 h à température ambiante dans un tampon de blocage (5% BSA dans du TBS-T, sature les sites non spécifiques sur lesquels l’anticorps pourrait s’accrocher) sous agitation. Ensuite, la membrane a été incubée pendant la nuit dans le tampon de blocage avec un anticorps primaire à la concentration souhaitée à 4°C sous agitation. La membrane a été lavée 3 × 5 min dans du TBS-T et incubée dans le tampon de blocage avec l’anticorps secondaire reconnaissant l’anticorps primaire pendant 1 h à température ambiante. Un ECL a été réalisé. Les images ont été prises par ImageQuant LAS500 (GE Healthcare, Life Sciences, Chicago, IL, USA) et l’intensité relative des bandes de protéines a été quantifiée à l’aide d’ImageJ.
Un exemple de méthode de quantification relative du signal sur western-blot :
Immunoprécipitation
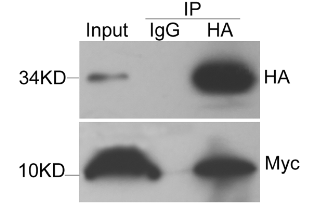
Cette technique permet d’isoler une protéine d’un extrait cellulaire et tous les interactants directs ou indirects avec cette protéine grâce à l’interaction très spécifique anticorps-antigène. Les anticorps peuvent être fixés sur des billes et on peut récupérer dans le culot les complexes formés après une centrifugation. Alternativement, on peut utiliser des billes métalliques qui sont ensuite attirées par un aimant. L’enrichissement de la fraction immunoprécipitée par rapport à la fraction initiale (souvent appelée input) peut être appréciée par western-blot.
Cette technique permet de savoir si deux protéines A et B appartiennent à un même complexe à un moment donné et dans des conditions données. On fait une immunoprécipitation avec un anticorps contre A et on analyse la présence par western-blot de la protéine B reconnue par un autre anticorps. Pour valider complètement l’interaction, il est préférable aussi de montrer l’inverse (immunoprécipitation de B et révélation de A).
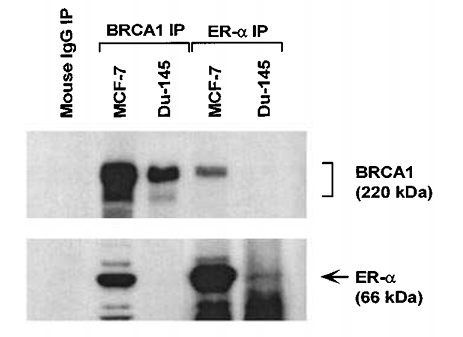
Les protéines récupérées lors d’une immunoprécipitation peuvent aussi être analysées de manière non biaisée par spectrométrie de masse.
Si de bons anticorps ne sont pas disponibles contre une protéine donnée pour faire l’immunoprécipitation, on peut faire exprimer via des vecteurs d’expression dans les cellules ou par transgénèse dans l’embryon des protéines dites taguées avec quelques acides aminés supplémentaires qui sont reconnus par un anticorps spécifique (tag HA, tag Myc ou tag FLAG par exemple).
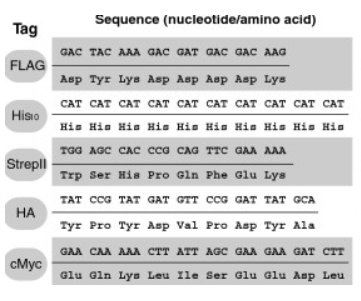
Le double-hybride
Le système du double hybride effectué chez la levure ou dans des cellules de mammifères permet de mettre en évidence l’interaction entre deux protéines. Il est basé sur le nature modulaire des facteurs de transcription qui sont généralement composés de deux domaines : un domaine de liaison à l’ADN et un domaine d’activation de la transcription. Chez la levure, on utilise le facteur de transcription Gal4. Si on veut savoir si la protéine A interagit avec la protéine B, on fait synthétiser aux cellules deux protéines de fusion : une avec le domaine de liaison à l’ADN de Gal4 fusionné avec la protéine A et une autre avec le domaine d’activation de Gal4 fusionné avec la protéine B. Ce n’est seulement dans le cas où A interagit avec B que le domaine d’activation de la transcription se trouvera à proximité du bon domaine de liaison à l’ADN et alors la transcription d’un gène rapporteur (codant la luciférase, la GFP ou une protéine indispensable à la croissance des colonies de levure) sera activée.
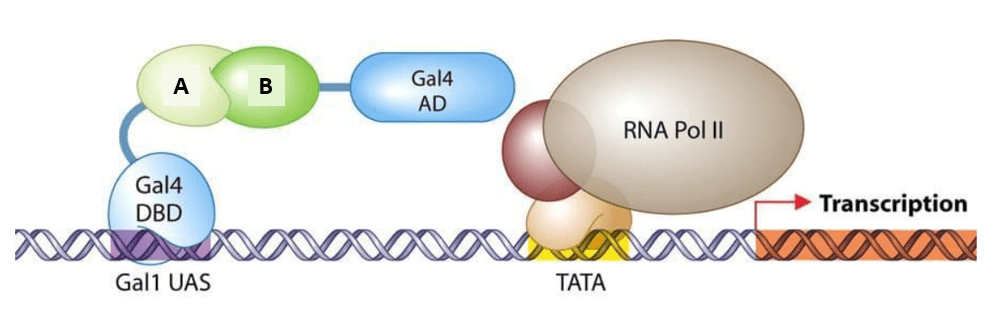
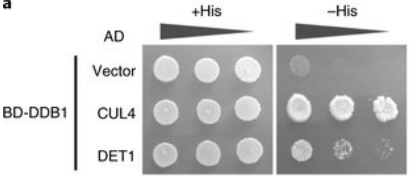
On peut utiliser un système équivalent dans des cellules de mammifères ce qui apporte l’avantage que les protéines testées pour l’interaction sont dans un environnement plus « physiologique » que lorsqu’elles sont exprimées dans la levure.
Immunohistochimie/Immunocytochimie/Immunofluorescence
Comme le western-blot, ces méthodes utilisent la spécificité de reconnaissance des anticorps pour mettre en évidence les protéines d’intérêt mais cette fois-ci non pas à partir d’extraits protéiques mais dans des cellules (cyto) ou des tissus (histo) qui ont été fixés au préalable. Ainsi, il y a une résolution spatiale qui est nettement plus précise.
Un exemple d’étude immunocytochimique :
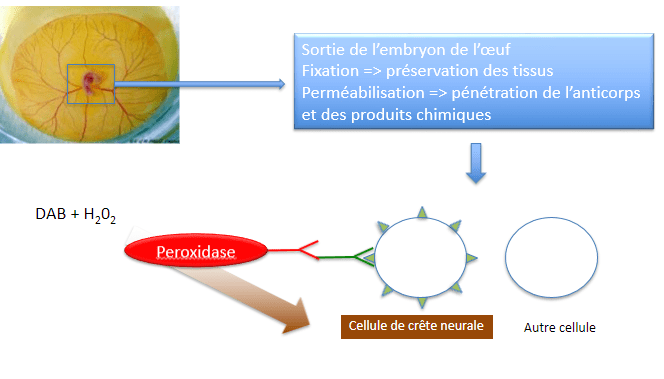
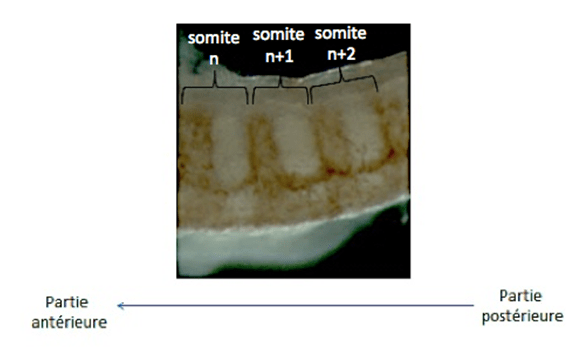
L’immunochimie est basée sur la révélation d’une activité enzymatique apportée par l’anticorps secondaire alors que l’immunofluorescence est basée sur l’émission de photons à une longueur d’onde donnée d’un fluorophore couplé à l’anticorps secondaire.

On peut faire plusieurs détections d’immunofluorescence sur un même échantillon à condition de bien choisir l’origine des anticorps (pas d’espèces communes pour la production des différents anticorps primaires) et les longueurs d’onde des fluorophores des anticorps secondaires.
L’immunohistochimie ou l’immunohistofluorescence peuvent se faire sur des embryons entiers ou plus souvent sur des coupes histologiques. Pour réaliser ces coupes, les échantillons fixés sont inclus dans de la paraffine pour les maintenir et sont coupés au microtome.

Alternativement, les échantillons peuvent être inclus dans de l’OCT (un mélange d’alcool polyvynilique et de polyéthylène glycol) et coupés à -20°C dans un cryostat.

Les progrès de la microscopie et de l’analyse des images
Généralités
Les progrès scientifiques sont indissociables des progrès techniques et cela s’applique tout particulièrement à la microscopie. L’observation des cellules a bénéficié des progrès de l’optique, puis des progrès chimiques (pour les colorants), des progrès de la génétique (pour créer et introduire dans les cellules des protéines-fusion avec des protéines fluorescentes telles que la GFP), et des progrès de l’informatique (pour traiter les images).
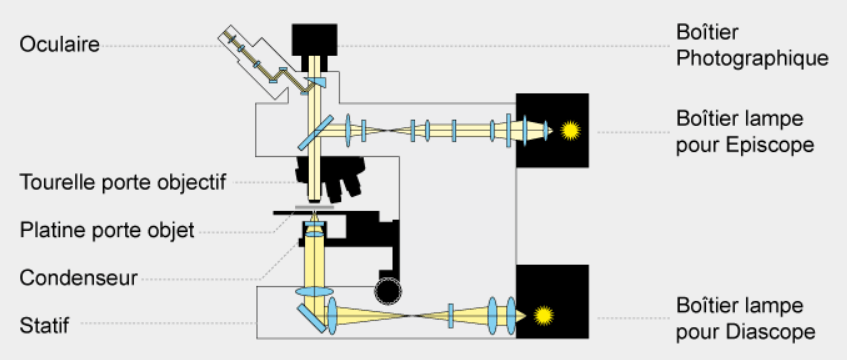
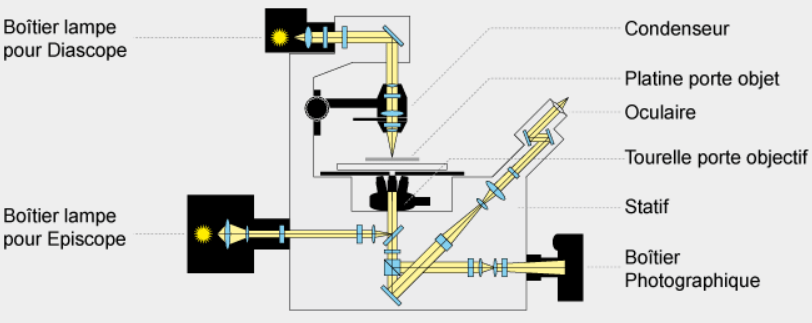
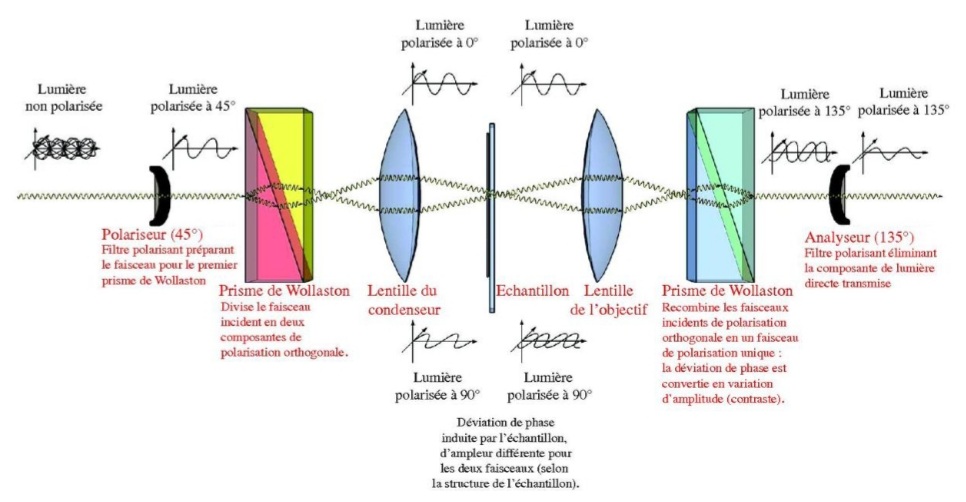
La résolution d’un microscope photonique (la distance à partir de laquelle on peut distinguer deux points) peut descendre jusqu’à 0,2 µm. Des objets plus petits et brillants peuvent être aperçus mais à cause d’effets de diffraction, ils apparaissent flous (selon la fonction d’étalement du point). Lorsque la lumière traverse un objet complexe les phases de ses ondes peuvent se décaler en fonction de l’indice de réfraction du milieu qu’elles traversent. C’est cette propriété qui est exploitée par le microscope à contraste de phase. Cet effet est amplifié par le microscope à contraste interférentiel (ou microscope Nomarski). Le faisceau lumineux est dédoublé en 2 faisceaux proches l’un de l’autre par un prisme avant la traversée de l’objet. Après la traversée, un second prisme recompose les 2 faisceaux qui produisent des interférences qui dépendent de l’épaisseur et de la biréfringence des objets observés. Ce type de microscopie accentue les contrastes au bord des structures.
En microscopie « classique » l’image nette de l’échantillon qui se trouve dans le plan focal peut être noyée dans les images floues qui proviennent de l’échantillon au-dessus ou en dessous de ce plan. Le microscope confocal permet d’éviter cet inconvénient et permet de réaliser des images nettes de très faible profondeur de champ (environ 400 nm). On peut parler de section optique. C’est réalisé grâce à la présence d’un petit orifice (ou sténopé) devant le détecteur dans un plan focal conjugué au plan focal de l’objectif. Il fera office de « filtre optique » ne laissant passer vers le détecteur que les photons provenant du plan de l’échantillon où l’image sera nette.
La manière dont on illumine les échantillons a aussi fait des progrès. Pour les études en fluorescence on utilise des lasers argon-ion (pour 457 nm, 488 nm, 514 nm) et hélium-néon (pour 543 nm, 633 nm).
L’angle avec lequel on illumine l’échantillon a aussi son importance. La microscopie TIRF (pour Total Internal ReFlection microscopy) permet de ne juste voir que les molécules fluorescentes à la membrane plasmique ou juste en dessous en utilisant un angle d’incidence particulier de la lumière d’excitation de la fluorescence.
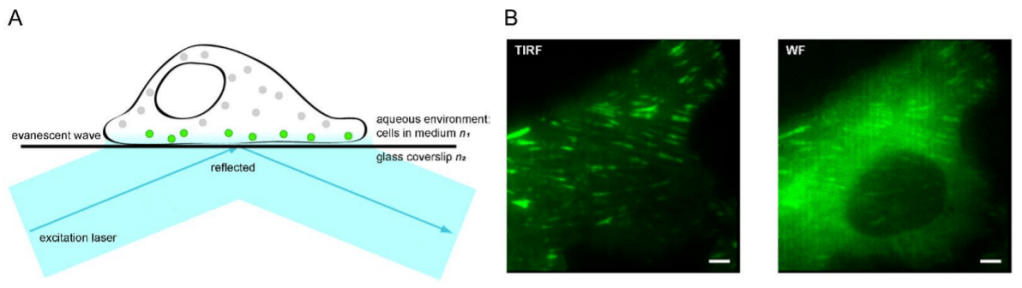
La réflexion de la lumière laser produit un champ électromagnétique connu sous le nom d’onde évanescente, qui diminue de façon exponentielle. Il est
seulement assez puissant pour exciter les fluorophores (cercles verts) dans une couche mince d’environ 100-200 nm au-dessus de la lamelle, laissant
les fluorophores à d’autres emplacements cellulaires non excités (cercles gris). On peut ainsi observer la fluorescence uniquement à la membrane plasmique ou juste en dessous (B) Le même champ de vision, pris en image en microscopie TIRF (à gauche) ou en microscopie normale (à droite), d’une cellule d’ostéosarcome exprimant paxilline-GFP. Barre d’échelle = 2 µm. Source : https://www.mdpi.com/2079-7737/10/11/1189
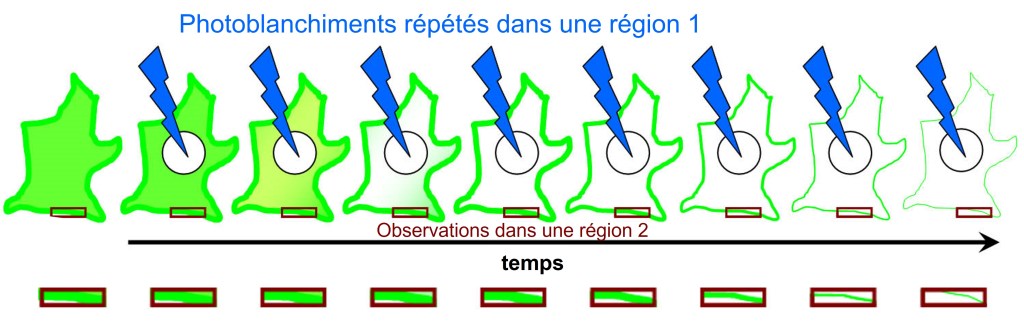
Le FRAP (Redistribution de fluorescence après photoblanchiment) et le FLIP
Le photoblanchiment de la fluorescence est habituellement un problème lors de l’observation d’échantillons contenant des fluorophores. Néanmoins, on peut en tirer avantage en épuisant la fluorescence dans une région bien précise de la cellule et en observant la cinétique avec laquelle la fluorescence est restaurée dans celle-ci. La restauration proviendra forcément de l’arrivée de nouvelles molécules fluorescentes et ainsi la dynamique de diffusion ou de transports de molécules marquées avec un fluorophore peut être étudiée.
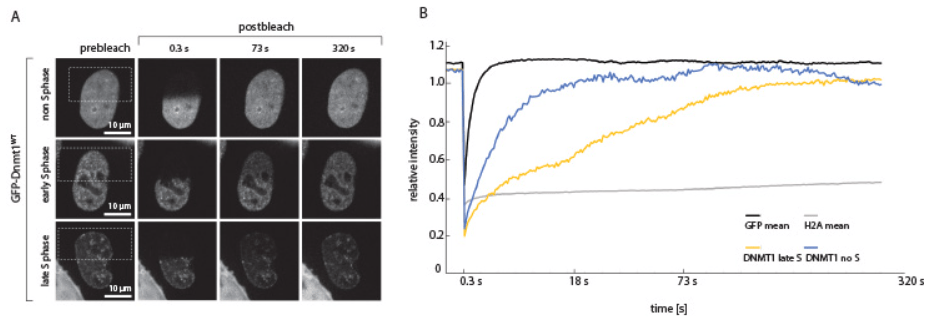
Le FLIP (ou perte de fluorescence induite par photoblanchiment) suit le principe inverse : on photoblanchie de multiples fois une zone où des protéines marquées avec un fluorochrome sont présentes et on étudie la diminution de la fluorescence ailleurs dans la cellule.

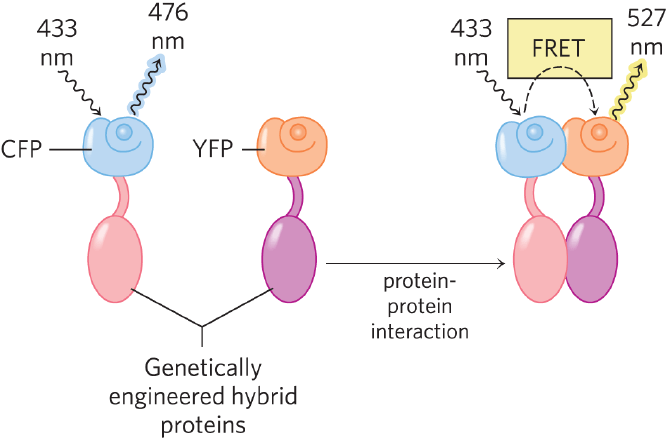
Le FRET (Transfert d’énergie entre molécules fluorescentes)
Cette technique permet de voir en microscopie si deux fluorophores se trouvent à moins de 10 nm l’un de l’autre. Pour ce faire, la longueur d’émission d’un fluorophore A doit pouvoir exciter un fluorophore B dont on pourra enregistrer l’émission de photons. Lorsque chaque fluorophore est porté par une protéine différente et qu’un signal FRET est observé, on peut en déduire que les deux protéines se trouvent en étroite proximité et qu’elles interagissent entre elles. Comme couple de fluorophore utilisé, on peut citer le CFP comme transmetteur d’excitation au YFP.
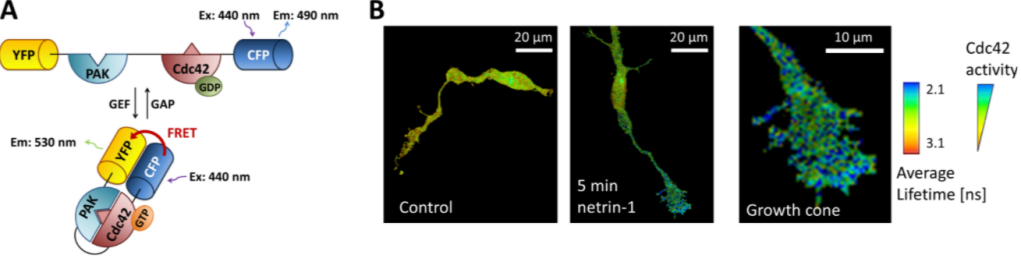
On peut aussi construire des biosenseurs basés sur le FRET comme dans les exemples qui suivent :

Approches optogénétiques pour perturber la signalisation cellulaire
L’optogénétique est un domaine en plein essor où la lumière à une longueur d’onde précise agit sur des protéines et change leur conformation, ce qui provoque des modifications dans la physiologie cellulaire. En neurosciences, l’ouverture ou la fermeture par la lumière de canaux ioniques permet de manipuler les dépolarisations des cellules nerveuses. Pour l’étude de la signalisation cellulaire, CRY2 (le cryptochrome 2 de la plante Arabidopsis) s’avère un outil de choix par sa capacité à s’homo-oligomériser lorsqu’il est stimulé par la lumière bleu ou à s’hétéro-dimériser avec la protéine CIB1 (Duan et al., 2017). Ainsi, à l’aide de protéines-fusion contenant CRY2 ou CIB1, on peut activer par la lumière une interaction.
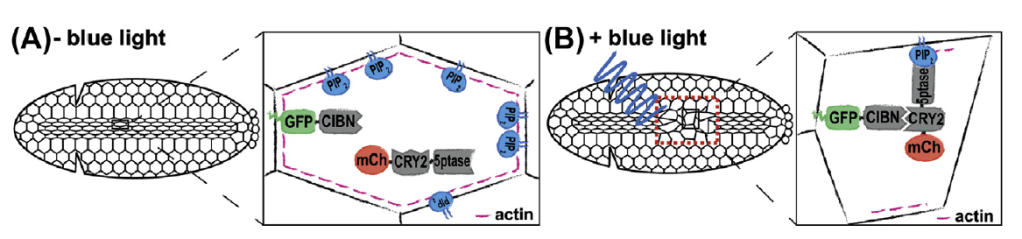
De nombreuses possibilités et combinaisons sont ainsi offertes pour contrôler l’activité d’une protéine et donc étudier le mécanisme de nombreux processus cellulaires du développement.

Les rapporteurs fluorescents pour la concentration calcique
Les concentrations cytoplasmiques de Ca2+ sont habituellement basses et l’augmentation de cette concentration par l’ouverture de canaux du réticulum endoplasmique ou de la membrane plasmique augmente l’activité de certaines protéines et déclenche des cascades de signalisation.
Des molécules à la fluorescence augmentée par les ions Ca2+ sont devenus des outils précieux pour ce domaine de recherche. La première molécule a avoir été populaire dans les laboratoires est le Fura-2, un acide aminopolycarboxylique, découvert en 1986. Il absorbe à 340-380 nm (UV) et son émission à 510 nm dépend de la concentration calcique.
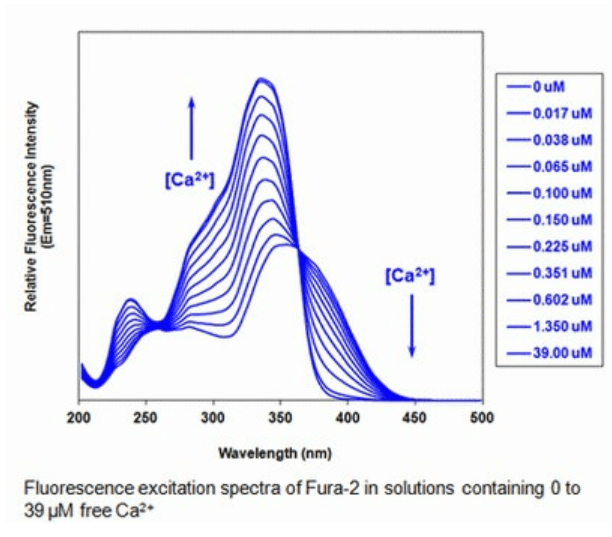
Plus récemment, une protéine fusion entre la GFP, la calmoduline (une molécule de signalisation sensible à la concentration en Ca2+) et M13 (une courte séquence de la kinase de la chaine légère de la myosine) a été mise au point, et s’appelle GCamp.
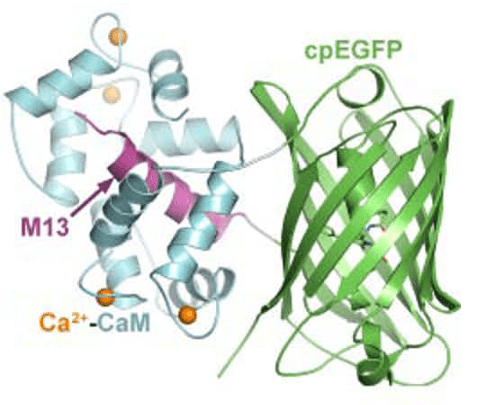
Comme c’est une protéine, on peut la faire exprimer après transfection ou transgénèse de l’ADN correspondant (et grâce à un promoteur spécifique par exemple) dans les cellules ou les embryons.
La cytométrie de flux
Il s’agit de marquer un ou plusieurs élément.s cellulaire.s d’intérêt avec un fluorochrome (fluorochrome lié à un anticorps ou molécule rendue fluorescente quand elle se lie à l’ADN par exemple) puis de séparer les cellules pour les faire passer une à une sous un laser à la longueur d’onde d’excitation de la fluorescence à observer. Un comptage du nombre de cellules en fonction de l’intensité de fluorescence est réalisé. Il peut y avoir un tri des cellules selon leur niveau de fluorescence, le flux cellulaire pouvant être automatiquement dévié vers différents tubes en fonction des fluorescences mesurées. Dans ce cas-là, on parle de FACS (Fluorescent Activated Cell Sorting), même si le terme de FACS s’est étendu à la méthode où on ne fait que compter les cellules.
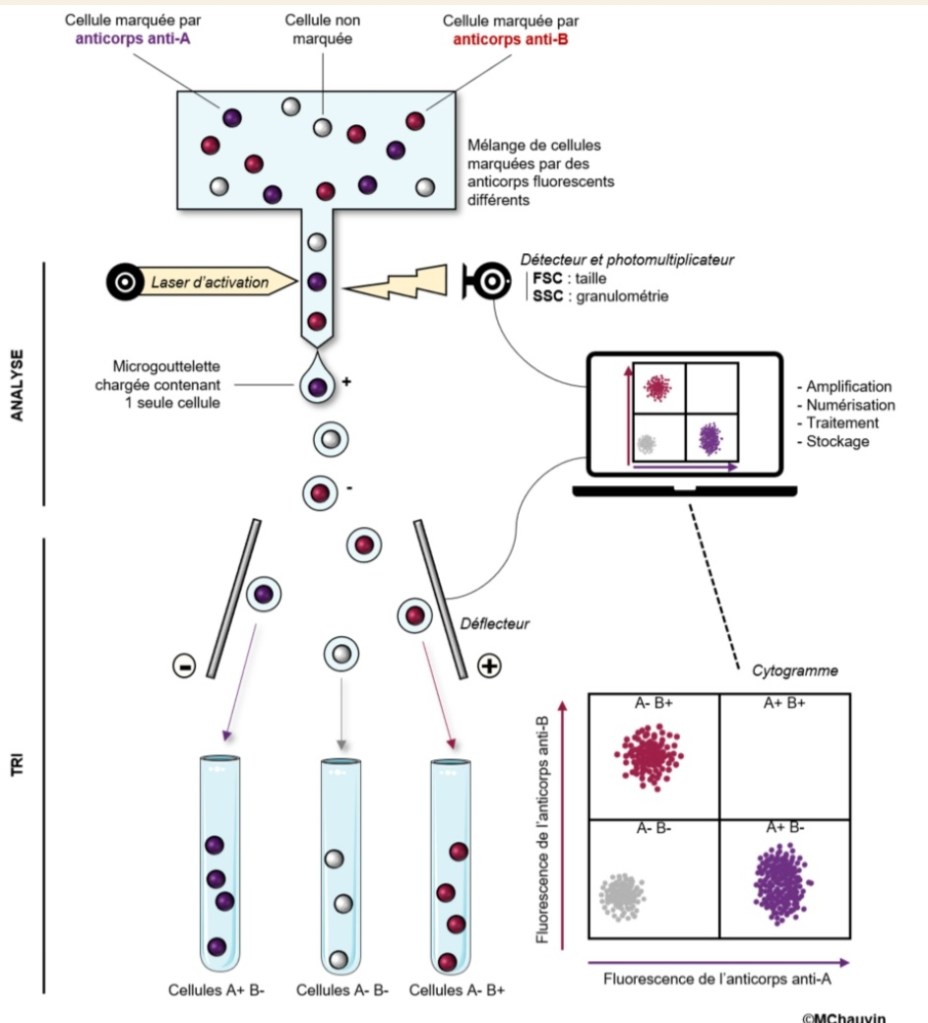
Exemple d’utilisation :
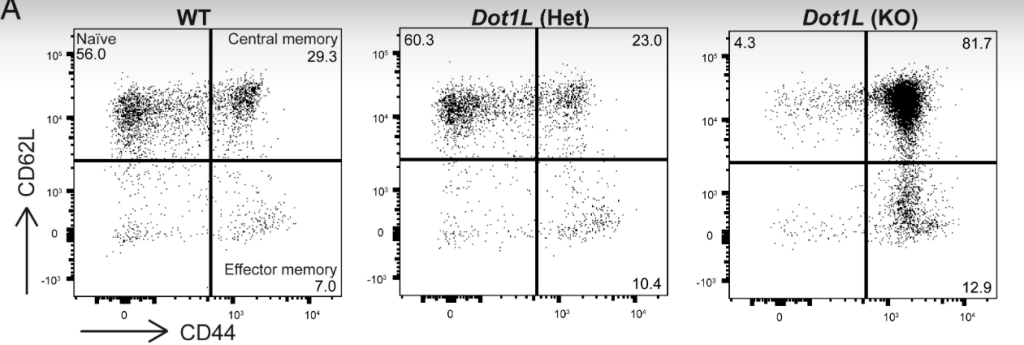
La microscopie électronique
L’illumination de l’échantillon se fait avec un faisceau d’électrons et non pas un faisceau de photons, contrairement à la microscopie photonique. Les microscopes électroniques ont un pouvoir de résolution près de 1000 fois supérieur à celui des microscopes photoniques (0,2 nm contre 0,2 µm) car la longueur d’onde de De Broglie d’un électron est bien plus petite que la longueur d’onde d’un photon, même dans les UV. Les lentilles utilisées ne sont plus optiques mais électromagnétiques. La fixation de l’échantillon est le plus souvent réalisée par du glutaraldéhyde (qui ponte les protéines) puis du tétroxyde d’osmium (qui réagit avec les lipides et notamment les phospholipides des membranes biologiques, ce qui les rend plus visibles). Dans le cas particulier du microscope électronique à balayage, un faisceau d’électron très fin balaie la surface d’un échantillon qui réémet en réponse des électrons qui sont analysées et une image en 3D de l’objet observé peut être réalisée.
Les tests de migration cellulaire
Test de cicatrisation
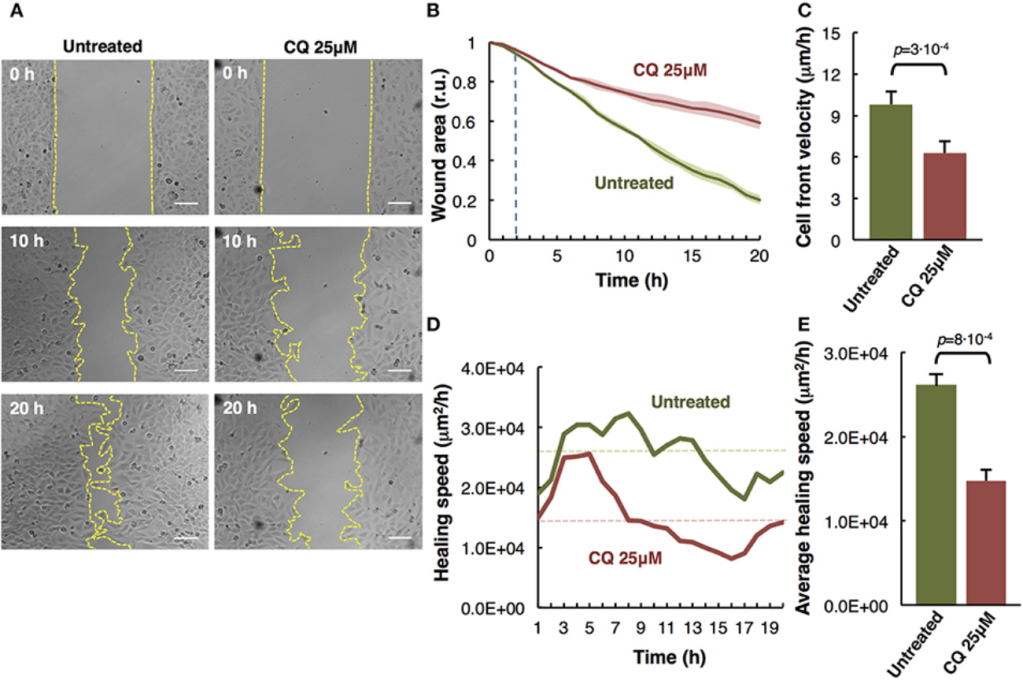
Le test de cicatrisation liée à la blessure de tapis cellulaire consiste à arracher les cellules de manière contrôlée sur une trajectoire précise dans une boîte de Pétri et de filmer la manière dont les cellules avoisinantes de la « blessure » vont coloniser l’espace vide.
La principale composante de la cicatrisation de la blessure est la migration cellulaire mais il peut y avoir aussi une composante liée à la prolifération. Egalement, la matrice extracellulaire peut être abimée lors de la réalisation de la blessure.
Test de migration en chambre de Boyden
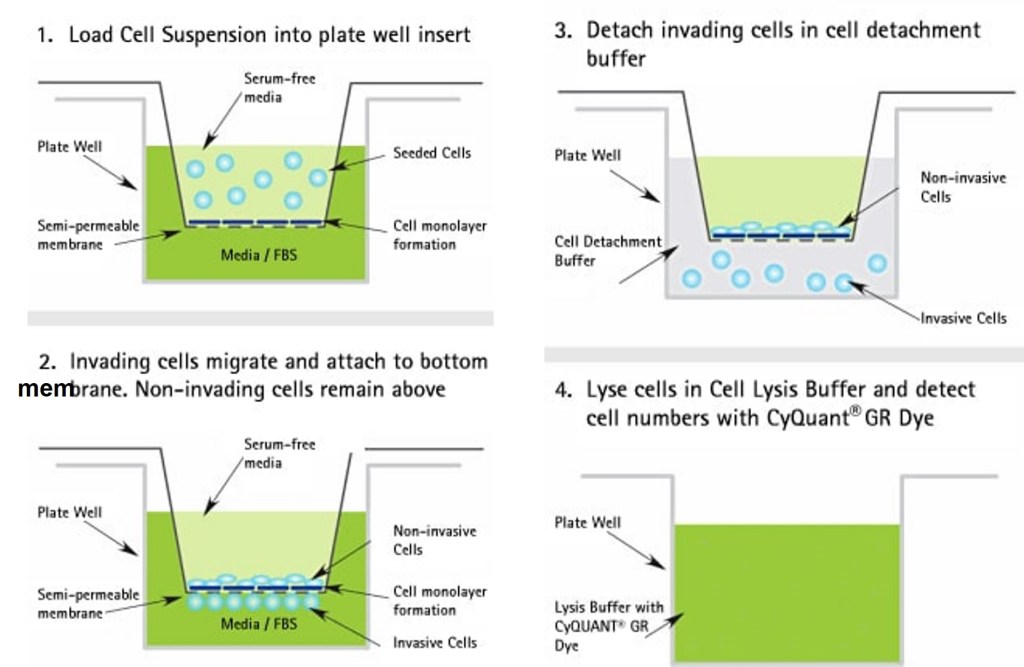
Ce test repose sur deux compartiments avec des milieux de composition différente séparés par une membrane poreuse (en général avec des pores d’un diamètre entre 3 et 12 µm (à adapter selon les cellules étudiées)). Les cellules sont déposées sur cette membrane et on les incube durant une durée permettant à certaines d’entre elles de traverser la membrane et de se retrouver à sa face inférieure dans le second compartiment. Celui-ci peut par exemple contenir un milieu avec un chémoattractant.
Les marqueurs de prolifération
BrdU

Il s’agit d’un analogue de nucléoside qui a une structure similaire à la thymine (avec un brome à la place d’un -CH3). Lorsqu’on met des cellules dans un milieu avec du BrdU ou qu’on l’injecte dans la circulation sanguine d’un embryon, il rentre dans les cellules et s’incorpore dans l’ADN uniquement s’il y a réplication. On replace ensuite les cellules dans un milieu sans BrdU (et il est dégradé rapidement dans un organisme). La population de cellules qui a répliqué son ADN lors de sa présence reste marquée et même si elle prolifère encore par la suite ou migre ou se différencie, on pourra toujours détecter la présence de BrdU avec un anticorps spécifique et les techniques habituelles d’immunomarquage. Cette technique permet donc un marquage permanent d’une population de cellules qui a proliféré à un moment donné.
Ki-67 et PCNA
Pour suivre la prolifération dans un tissu, on peut également faire des immunomarquages contre des protéines qui ne sont présentes que lors des phases G1 à M mais pas en phase G0.
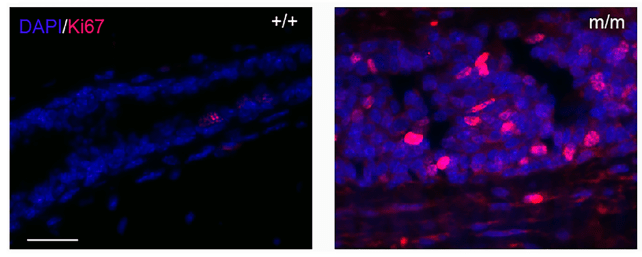
- Ki-67 est une protéine nucléaire impliquée dans la transcription des ARN ribosomaux. Elle est exprimée tout au long du cycle de G1 à M mais est absente des cellules en G0. Durant la mitose où il n’y a plus de noyaux, Ki-67 est localisée sur les chromosomes. La protéine est nommée d’après l’anticorps dont elle s’est avérée l’antigène par la suite. L’anticorps a été produit dans la ville allemande de Kiel (d’où le « Ki ») et était en position 67 dans une plaque de 96 puits avec divers anticorps produits contre des noyaux de lymphomes de Hodgkin.
- PCNA est un facteur de processivité pour l’ADN polymérase lors de la réplication et est aussi impliqué dans la réparation de l’ADN. Son expression est très faible dans des cellules quiescentes et est fortement activée lors du passage G1/S.
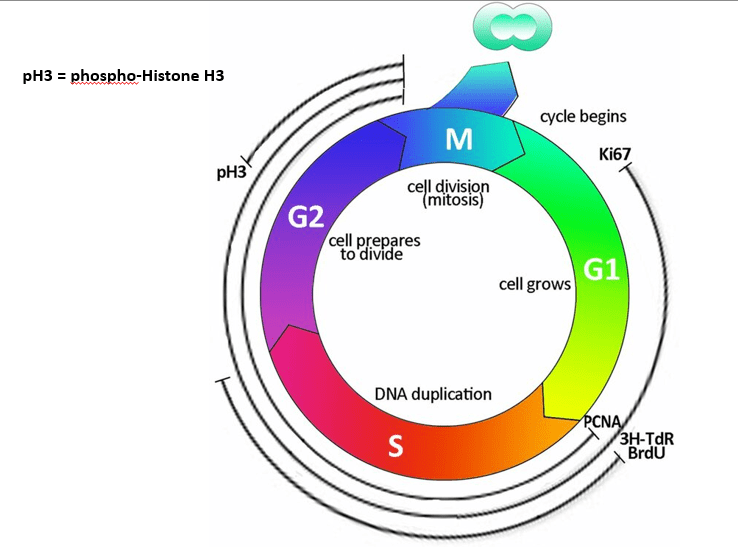
L’étude du cycle cellulaire
Utilisation du FACS
La quantité d’ADN varie au cours du cycle cellulaire (elle est doublée durant la phase S et revient à la valeur habituelle lorsque les cellules se séparent en fin de mitose). Avec un fluorophore dont la fluorescence dépend de l’ADN, on peut utiliser le tri cellulaire contrôlé par fluorescence (FACS) pour déterminer dans une population cellulaire la proportion des cellules dans différentes phases du cycle.

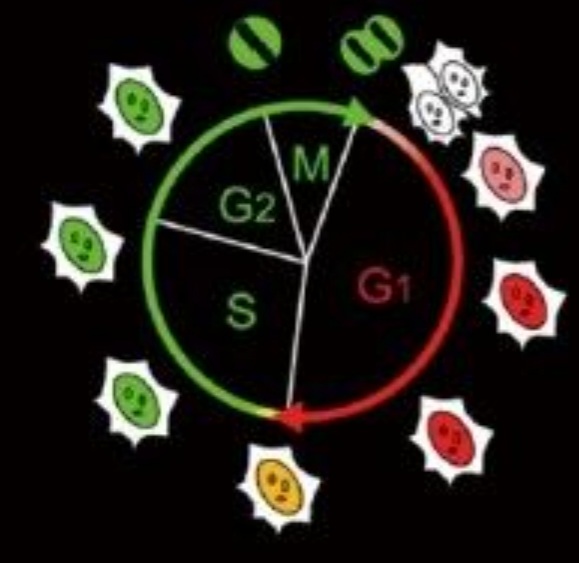
Le système FUCCI
FUCCI (Fluorescent Ubiquitination-based Cell Cycle Indicator) est un ensemble de sondes fluorescentes qui permet de visualiser la progression du cycle cellulaire en temps réel dans des cellules vivantes. FUCCI utilise l’expression des protéines Cdt1 et Geminin qui dépend de la phase du cycle cellulaire. Une protéine de fusion d’un fragment de Cdt1 (acides aminés 30-120) avec la protéine fluorescente Kusabira-Orange 2 (mKO2) sert d’indicateur de la phase G1. Une protéine de fusion d’un fragment de Geminin (acides aminés 1-110 ou 1-60) avec la protéine fluorescente Azami-Green 1 (mAG1) permet de visualiser les phases S, G2 et M.
DES OUTILS POUR ANALYSER/MONTER DES IMAGES EN BIOLOGIE CELLULAIRE
L’application libre Fiji (ou ImageJ) . Lien vers des tutoriels pour utiliser Fiji (en anglais).
L’application libre Cell Profiler
AUTRES LIENS SUR LES TECHNIQUES ET OUTILS POUR LA BIOLOGIE CELLULAIRE :
Sur les différents types de microscopie : https://micro.magnet.fsu.edu/
EN DIRECT DES LABOS :
- Adhérences cellule-cellule
- Arabidopsis thaliana
- Axe antéro-postérieur chez la drosophile
- Biomécanique du développement
- Caenorhabditis elegans
- Concepts principaux
- Contrôle de la traduction
- Contrôle de la transcription
- Contrôle génétique et épigénétique
- Croissance du tube pollinique et double fécondation chez les Angiospermes
- Croissance et guidage axonal
- Des modèles animaux moins classiques
- Développement de l’oeil des Vertébrés
- Développement et évolution
- Et l’Humain ?
- Exercices sur l’ovogenèse, la spermatogenèse et la fécondation
- Exercices sur le contrôle de l’expression des gènes
- Exercices sur le développement des bourgeons de membre
- Exercices sur le développement des muscles striés squelettiques
- Exercices sur le développement des végétaux et les hormones végétales
- Exercices sur les cycles et les divisions cellulaires
- Exercices sur les étapes du développement, les inductions embryonnaires et la mise en place des axes de polarité
- Exercices sur les matrices extracellulaires, le cytosquelette et les adhérences cellule-cellule
- Exercices sur les voies de signalisation
- Glossaire
- Glossaire des termes liés à la génétique
- Glossaire des termes liés au cytosquelette, la matrice extracellulaire, l’adhérence et la migration cellulaire
- Hématopoïèse et développement des cellules du système immunitaire
- Histoire de la biologie cellulaire et de la biologie du développement
- L’acide rétinoïque
- L’apoptose
- L’autophagie
- L’organogenèse
- L’ovogénèse prépare le développement embryonnaire
- La drosophile
- La fécondation
- La formation des somites
- La gastrulation
- La gastrulation (version allégée)
- La métamorphose chez les Hexapodes et les Amphibiens
- La neurogénèse chez les mammifères adultes
- La neurulation
- La poule
- La signalisation calcique
- La souris
- La superfamille TGFβ et ses voies de signalisation
- La voie de signalisation de l’auxine et ses rôles
- La voie de signalisation Hedgehog
- La voie de signalisation Hippo et ses composants YAP/TAZ
- La voie de signalisation Notch
- Le clivage
- Le cytosquelette
- Le destin des cellules et les réseaux de régulation génique
- Le développement des bourgeons de membre
- Le développement des muscles striés squelettiques
- Le développement des organes génitaux et des cellules germinales
- Le développement du cortex
- Le méristème apical caulinaire en phase végétative et lors de la formation d’une fleur
- Le poisson zèbre
- Le xénope
- Les cellules des crêtes neurales
- Les cellules et les gènes en action dans le développement
- Les cellules souches
- Les cellules tumorales
- Les cycles et les divisions cellulaires
- Les étapes du développement
- Les étapes du développement embryonnaire d’Arabidopsis thaliana et leur contrôle
- Les inductions embryonnaires et les gradients de morphogène
- Les matrices extracellulaires animales
- Les organismes modèles
- Les outils pour étudier l’expression et la fonction des gènes
- Les parois des cellules végétales
- Les transitions épithélio-mésenchymateuses et les migrations cellulaires
- Les vésicules extracellulaires
- Les voies de signalisation
- Les voies de signalisation FGF
- Mise en place des axes chez les Vertébrés
- Structures et processus cellulaires
- Voies de signalisation WNT

{kind=link}
{kind=link}
{kind=link}