
Niveaux de difficulté : * = embryon; ** = têtard; *** = mature
- EXERCICE 1 sur les étapes du développement
- EXERCICE 2 sur le développement de la souris
- EXERCICE 3 sur la fécondation et l’appareil génital de la souris
- EXERCICE 4 sur la voie Wnt et le développement des crêtes neurales
- EXERCICE 5 sur l’embryon de poulet et les cellules de crêtes neurales
- EXERCICE 6 sur le développement précoce de l’embryon de xénope
- EXERCICE 7 sur la mise en place des axes du xénope
- EXERCICE 8 sur le développement du xénope
- EXERCICE 9 sur le développement précoce du xénope
- EXERCICE 10 sur la mise en place des axes du xénope et la voie Wnt
- EXERCICE 11 sur la mise en place des axes du poisson-zèbre
- EXERCICE 12 sur la mise en place des axes et des feuillets chez le poisson-zèbre
- EXERCICE 13 sur la mise en place des axes chez le xénope
- EXERCICE 14 sur la mise en place des axes chez le xénope
- EXERCICE 15 sur le développement des crêtes neurales
- EXERCICE 16 sur la mise en place des axes du xénope
- EXERCICE 17 sur le rôle des gènes Hox
- REPONSES
EXERCICE 1 sur les étapes du développement

*1.1 : Quel est l’organisme modèle utilisé et quelle(s) étape(s) du développement observe-t-on ? Quel pourrait être la taille correspondante à la barre d’échelle ? (estimation)
*1.2 : A quelle vue (axe) correspond la première photo ? Et la dernière ? De quel côté se déplace le blastopore ?
*1.3 : Comment s’appelle le mouvement qui provoque le changement de forme de l’embryon dans les dernières photos ?
EXERCICE 2 sur le développement de la souris

*2.1 : A E6,5, les embryons de souris sont-ils implantés dans l’utérus ?
*2.2 : A quelle étape du développement se trouve l’embryon de souris à E6,5 ?
*2.3 : Décrivez les coupes en A et B.
*2.4 : Quelles informations supplémentaires nous apporte la figure C ?
EXERCICE 3 sur la fécondation et l’appareil génital de la souris
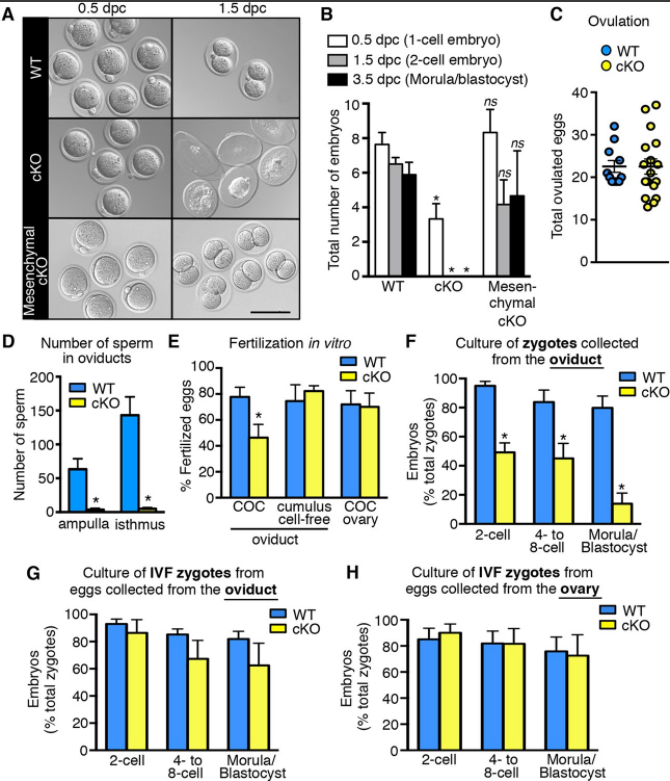
**3.1 : Comment peut-on réaliser une mutation conditionnelle du récepteur alpha aux œstrogènes soit dans l’épithélium de l’oviducte (cKO), soit dans le mésenchyme de l’oviducte ?
*3.2 A quoi voit-on qu’il y a bien eu fécondation à 0,5 dpc?
*3.3 Quel est le phénotype obtenu en A chez les mutants ?
*3.4 Que nous apprennent les figures C et D ?
**3.5 Que nous apporte comme informations la figure E ?
**3.6 Quelles conclusions peut-on tirer des données des figures F à H ?
EXERCICE 4 sur la voie Wnt et le développement des crêtes neurales

*4.1 Qu’est-ce qu’un dominant-négatif ?
*4.2 Qu’observe-t-on sur les coupes transversales ? Proposez des hypothèses pour expliquer le phénotype.
**4.3 Proposez des expériences pour valider ou invalider vos hypothèses.
*4.4 Que nous apprend le western-blot ? Quelle pourrait être la fonction de Cullin1 ?
**4.5 A l’aide de vos connaissances, essayez de trouver un scénario pour expliquer le phénotype.
**4.6 Sachant que la β-caténine joue un rôle essentiel dès le développement précoce du xénope, pourquoi l’effet du dominant-négatif de la Cullin1 n’est pas plus massif ?
EXERCICE 5 sur l’embryon de poulet et les cellules de crêtes neurales

**5.1 Combien de temps de développement sépare les embryons de 27 somites et de 30 somites ?
*5.2 Présentez la technique d’électroporation in ovo.
*5.3 De manière globale, quelles sont les cellules qui ont été électroporées ?
*5.4 Comparez les résultats des différentes électroporations réalisées à différents stades et tirez des conclusions concernant le développement des cellules de crêtes neurales.
EXERCICE 6 sur le développement précoce de l’embryon de xénope
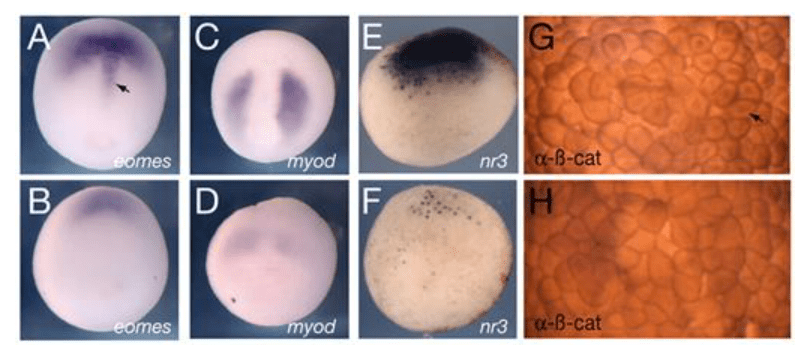
*6.1 Analysez et interprétez les résultats de l’étude de l’expression de eomes et myod dans les embryons issus d’ovocytes déplétés en Trim36.
**6.2 Analysez les résultats concernant l’expression de Xnr3 et précisez en quoi ils sont liés aux résultats de la question précédente.
*6.3 Analysez et interprétez les résultats de l’immunohistochimie anti-beta-caténine. Proposez des hypothèses concernant le rôle de Trim36.
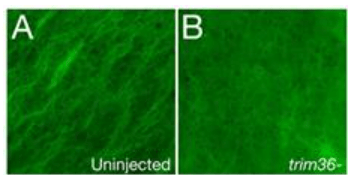
*6.4 Pourquoi cette observation est-elle faite 80 minutes après la fécondation ?
*6.5 Qu’observez vous et comment cela est-il lié aux résultats des questions précédentes ?
**6.6 Proposez une expérience qui pourrait sauver le phénotype des embryons (en dehors d’injecter une forme de l’ARNm Trim36 insensible au MO)
EXERCICE 7 sur la mise en place des axes du xénope

**7.1 Comment peut-on reconnaître sur des embryons de xénope au stade 4 cellules, les cellules ventrales et les cellules dorsales ?
*7.2 Analysez et interprétez ces résultats. Quel autre ARNm codant une autre protéine pourrait donner le même résultat après une injection similaire ?

*7.3 Que provoque l’irradiation aux UV ?
*7.4 Analysez et interprétez l’expérience en B.
*7.5 Quelles informations supplémentaires nous apporte l’expérience C ? Posez des hypothèses sur l’action de GBP.
**7.6 Des fonctions similaires à quelle autre protéine GBP pourrait-elle avoir ?
EXERCICE 8 sur le développement du xénope

*8.1 Qu’indique bc sur la légende de la figure A ? Et qu’indique la pointe de flèche blanche en B et C ?
*8.2 Analysez et interprétez les résultats de l’expérience.
EXERCICE 9 sur le développement précoce du xénope

*9.1 Comparez le résultat avec la calotte animale seule et lorsque la calotte animale est associée aux cellules de l’hémisphère végétatif (Con/Con). Expliquez ce qu’il s’est passé dans les agrégats.
*9.2 Que nous apprend sur VegT l’expérience dont le résultat est présenté dans le puits 5 ?
*9.3 Analysez et interprétez le rôle de Bix4.
EXERCICE 10 sur la mise en place des axes du xénope et la voie Wnt

*10.1 Quel est le phénotype des embryons où Wnt11 n’est pas produit ? Quel est l’intérêt de faire les co-injections de l’ARNm de Wnt1 avec l’ARN anti-sens Wnt11 ?
*10.2 Analysez et interprétez les résultats obtenus avec l’injection de l’ARNm de LRP6.
*10.3 Analysez et interprétez les résultats obtenus en C et D.
**10.4 Proposez un modèle pour expliquer les résultats.
EXERCICE 11 sur la mise en place des axes du poisson-zèbre

*11.1 Quelles sont les malformations observées en A ? Comment les interpréter ?
*11.2 Quelles informations supplémentaires nous apporte l’expérience avec l’injection d’ARNm codant Bmp2b co-injectés ou non avec des ARNm codant Bif1 ?

*11.3 Analysez et interprétez les résultats de cette expérience.
EXERCICE 12 sur la mise en place des axes et des feuillets chez le poisson-zèbre

*12.1 D’après les images et vos connaissances, dans quelle région est exprimée gsc ?
*12.2 Décrivez les effets des pertes-de-fonction de foxh1 et de eomesa.
*12.3 Que nous apprend la double perte-de-fonction foxh1 et eomesa ?
*12.4 Analysez et interprétez les résultats des injections d’ARNm de foxh1 dans un contexte sauvage ou mutant pour eomesa.
EXERCICE 13 sur la mise en place des axes chez le xénope

*13.1 Analysez et interprétez les données expérimentales concernant l’expression des marqueurs de foie et de poumon.
*13.2 Comment interpréter les résultats concernant l’expression des marqueurs de pancréas ?
EXERCICE 14 sur la mise en place des axes chez le xénope

*14.1 : Pourquoi l’expression d’un dominant-négatif des récepteurs aux BMP neuralise-t-il les calottes animales ?
*14.2 : Pourquoi utilise-t-on une calotte animale provenant d’un embryon pigmenté et une autre provenant d’un embryon non pigmenté pour les recombinaisons ?
**14.3 : Quel est l’effet de XMeis3 sur l’expression de Otx2, un marqueur de cerveau antérieur, dans les recombinants ?
**14.4 : Que peut-on déduire de l’étude de l’expression de Krox20, un marqueur de cerveau postérieur ?
EXERCICE 15 sur le développement des crêtes neurales

*15.1 Que se passe-t-il si on cultive les explants du stade 16 au stade 22 sans mésoderme et sans Edn1 ? Interprétez.
*15.2 Quel est le rôle du mésoderme intermédiaire mis en évidence ?
*15.3 Que peut-on conclure du rôle de la signalisation endothéline dans ce système avec ces expériences ?
EXERCICE 16 sur la mise en place des axes du xénope

*16.1 Comment peut-on savoir sur des embryons de xénope au stade 4 cellules où se développera la région ventrale ?
*16.2 D’après vos connaissances, où est exprimé l’ARNm de chordine ? Sachant que les embryons sur les photos sont tous orientés de la même manière déduisez où est exprimé wnt8.
*16.3 Analysez et interprétez l’effet de l’injection des ARNm codant Dscr6. A des stades plus tardifs, quel pourrait être l’effet sur l’organisation des embryons et des têtards ?
*16.4 Analysez et interprétez l’effet de l’injection conjointe des ARNm codant Dscr6 et Stat3C.
EXERCICE 17 sur le rôle des gènes Hox

**17.1 Interprétez les résultats avec le mutant MZ lzr.
**17.2 Que se passe-t-il si en plus on inhibe l’expression de pbx2 ?
**17.3 Qu’en déduisez vous sur le rôle des facteurs de transcription Hox dans le rhombencéphale ?
REPONSES
1.1 : Il s’agit d’un embryon d’Amphibien, sans doute un embryon de xénope. L’observation du blastopore indique que l’on observe la gastrulation et plus précisément sa partie terminale puisque le blastopore est déjà circulaire à la première image. A la dernière image, on voit l’embryon s’allonger selon un axe et la partie dorsale s’aplatir : on est au début de la neurulation. Un embryon de xénope fait à peu près 1 mm de diamètre donc la barre d’échelle doit correspondre à 500 µm. 1.2 : Il s’agit d’une vue du pôle végétatif sur la première photo (les macromères végétatifs forment le bouchon vitellin). Sur la dernière, on est en vue dorsale. Le blastopore se déplace du côté postérieur (il va former l’anus, on est chez un Deutérostomien). 1.3 : Des mouvements de convergence-extension.
2.1 : Oui, l’implantation a eu lieu à E4,75. 2.2 : Au début de la gastrulation. 2.3 : L’embryon muté est plus élargi que l’embryon sauvage avec les cellules du mésoderme qui sont moins jointives et qui forment un tissu plus lâche, voire lacunaire par endroit. L’endoderme est plus fin et discontinu. Une ligne primitive se forme tout de même dans la région postérieure et semble normale en comparaison avec l’embryon sauvage. 2.4 : En terme de morphologie générale, les différences se creusent entre le mutant et le sauvage à E8,5 par rapport à E6,5 avec l’embryon mutant qui est nettement plus petit ce qui n’était pas le cas deux jours auparavant. Brachyury est exprimé dans le mésoderme à ce stade et on constate que son expression est très faible chez le mutant, ce qui suggère un défaut majeur touchant les cellules du mésoderme, soit de détermination, soit de survie.
3.1 : On peut utiliser le système Cre-Lox avec la Cre sous le contrôle d’un promoteur spécifique de l’épithélium de l’oviducte ou spécifique du mésenchyme de l’oviducte et le gène codant le récepteur flanqué de deux séquences LoxP. 3.2 : Sur certains embryons, on voit clairement deux globules polaires. Le deuxième globule polaire n’est émis que juste après la fécondation, l’activation de l’ovocyte par l’entrée du spermatozoïde débloquant la méiose bloquée en métaphase II. 3.3 : Alors que les embryons semblent normaux à 0,5 dpc, les embryons portés par les mères cKO sont en train de dégénérer. 3.4 : Alors qu’il n’y a pas de différences de nombre d’ovocytes ovulés entre le sauvage et le mutant, le nombre de spermatozoïdes est très bas chez le mutant dans les différentes régions de l’oviducte. Le récepteur alpha aux œstrogènes est nécessaire dans l’épithélium de l’oviducte pour que les spermatozoïdes puissent y entrer ou y survivre. Ce n’est pas un problème de chemoattraction envers les ovocytes puisque ceux-ci sont présents en quantité normale. 3.5 Les COC prélevés dans les oviductes ne sont pas aussi aptes à être fécondés s’ils proviennent du mutant par rapport à s’ils proviennent du sauvage. Cela est dû à un défaut dans les cellules du cumulus oophorus car si on les enlève, le taux de fécondation redevient le même que chez le sauvage. Les COC provenant de l’ovaire se comportent normalement même en provenance du mutant. L’expression du récepteur alpha aux œstrogènes dans l’épithélium de l’oviducte est nécessaire pour que les cellules du cumulus oophorus soient pleinement fonctionnels. 3.6 Même dans des conditions de culture in vitro, les embryons issus de zygotes prélevés chez les mères mutantes ont une mortalité importante avec 2 paliers de mortalité : entre le zygote et le stade 2 cellules, et entre le stade 4/8 cellules et morula/blastocyste. Par contre, si on cultive des zygotes fécondés in vitro, on obtient des taux de survie normaux. On en conclut que la fonction du récepteur aux œstrogènes dans l’épithélium de l’oviducte est essentielle au moment de la fécondation via notamment une action sur les cellules du cumulus oophorus (voir les questions précédentes).
4.1 : Un dominant-négatif est une protéine (ou un gène muté codant une protéine) qui non seulement est inactive mais aussi empêche les protéines normales d’agir. 4.2 : On observe nettement plus de cellules pigmentées (des mélanocytes) du côté injecté que du côté contrôle. Cela s’accompagne de la quasi-disparition de l’expression de la N-tubuline hors du tube neural vue par hybridation in situ. Dans cette région, il est probable que les neurones soient des dérivés des cellules de crêtes neurales (CCN) tout comme les mélanocytes. L’expression du dominant-négatif de la Cullin1 a donc pu changer la détermination des dérivés des CCN, les faisant prendre un destin mélanocytaire plutôt que neuronal. On peut aussi imaginer que le dominant-négatif a fait proliférer les mélanocytes et a tué les neurones par apoptose. 4.3 : Il faudrait faire un marquage au BrdU (ou faire un immunomarquage Ki67) pour observer la prolifération et un marquage TUNEL pour observer l’apoptose. Pour l’hypothèse de la détermination, il faudrait faire des études in vitro sur des cultures de CCN isolées ou faire des études de marquage in vivo et suivre la destinée des cellules. 4.4 : La quantité de β-caténine est plus élevée en présence du dominant-négatif, ce qui veut dire que la Cullin1 inhibe l’expression de la β-caténine. Au vu de sa fonction d’E3 ubiquitine ligase, on peut supposer qu’elle ubiquitinyle la β-caténine et provoque sa dégradation. 4.5 : La β-caténine intervient dans la voie Wnt canonique. Or l’activation de cette voie oriente la détermination des CCN vers la voie mélanocytaire. Par conséquent, la plus faible dégradation de la β-caténine en présence du dominant-négatif de la Cullin1 pousse les CCN vers la voie mélanocytaire au détriment de la voie neuronale. 4.6 : Le dominant-négatif de la Cullin1 ne peut agir qu’en empêchant la fonction de la Cullin1. Si la Cullin1 n’est pas exprimée au cours du développement précoce du xénope, le dominant-négatif ne peut pas modifier quoi que ce soit au niveau d’expression de la β-caténine et son effet ne se fera ressentir que plus tard.
5.1 : Chez l’embryon de poulet, une paire de somites est générée toutes les 90 minutes donc les embryons à 30 somites sont (3x 1h30 =) 4h30 environ plus âgés que les embryons à 27 somites. 5.2 : On injecte une solution contenant un vecteur d’expression dans la lumière du tube neural puis on place 2 électrodes de part et d’autre de l’embryon. On fait passer de courts pulses de courant qui ouvrent les membranes et font rentrer l’ADN chargé négativement dans les cellules uniquement du côté de l’électrode positif (car l’ADN migre vers ce côté comme dans une électrophorèse). L’autre côté de l’embryon qui n’a pas reçu d’ADN sert de contrôle interne. 5.3 : Les cellules électroporées sont des cellules qui se trouvaient aux stades indiqués (15-25ss, 27ss…) d’un côté de la paroi du tube neural et à son sommet (qui correspond à l’ancienne bordure neurale). Certaines de ces cellules sont restées dans le tube neural, d’autres sont sorties du tube neural et expriment le marqueur HNK1, ce sont des cellules de crêtes neurales (CCN). 5.4 : Les CCN électroporées entre 15 et 25ss (c’est-à-dire marquées par la GFP depuis ce stade) donnent à E4,5, un ensemble assez large de dérivés : mélanoblastes, cellules du DRG, cellules de la racine ventrale, quelques cellules dans les ganglions sympathiques (GS). Les CCN marquées à 27 somites ne contribuent plus aux GS mais à tous les autres dérivés. A partir de 30 somites, les CCN marquées ne donnent plus que des mélanoblastes (quelques cellules dans les DRG à 30 somites mais nettement moins qu’à 27 somites). Quand on marque le tube neural au stade 40 somites, plus aucune CCN n’est produite. On en conclut qu’au cours du temps, la population de CCN dans le tube neural dorsal évolue et que globalement elle perd en multipotence pour devenir uniquement productrice de mélanoblastes aux stades tardifs. Notez qu’on raisonne sur une population et qu’on ne sait pas si même aux stades précoces, les CCN étaient chacune déjà unipotentes et que l’ensemble constituait un mélange hétérogène ou si les CCN étaient alors chacune multipotentes. L’émigration des CCN s’arrête entre les stades 40 et 44 somites.
6.1 : Chez les embryons affectés, eomes est exprimé mais plus faiblement que dans les embryons normaux. Il manque notamment une structure allongée dans le plan de symétrie bilatérale et c’est sans doute la corde (flèche noire). Le mésoderme paraxial et notamment les futurs muscles (myod) sont très affectés avec une expression très faible de myod. On en déduit que l’induction du mésoderme a eu lieu mais de manière nettement moins efficace qu’habituellement. 6.2 : On observe une expression très diminuée de Xnr3 dans les embryons issus des ovocytes sans Trim36. Xnr3 est exprimé dans la région dorsale par rapport au pôle végétatif et il donc il pourrait être exprimé dans le centre de Nieuwkoop et aussi dans l’organisateur de Spemann au vu de son extension dorsale. Les protéines Xnr sont impliqués dans l’induction de l’organisateur de Spemann et dans l’induction du mésoderme. C’est peut-être le défaut de Xnr3 qui est à l’origine des défauts mésodermiques (notamment dorsaux) observés à la question précédente. 6.3 : Dans l’embryon issu d’ovocytes normaux, on observe bien la β-caténine dans le noyau dans les cellules dorsales et cette localisation n’est plus présente (ou alors très faible) chez les embryons issus d’ovocytes sans Trim36. La localisation nucléaire de la β-caténine est essentielle pour la mise en place de l’organisateur de Spemann et les défauts observés sur des embryons plus âgés sont les conséquences de ce qui est observé ici. La localisation nucléaire de la β-caténine dépend du transport de Dishevelled au futur côté dorsal de l’embryon lors de la rotation corticale et on peut imaginer que Trim36 est nécessaire pour cet évènement. On peut aussi imaginer que Trim36 est nécessaire pour empêcher la dégradation de la β-caténine. 6.4 : La rotation corticale a lieu juste après (vers 1h30 après la fécondation pour des zygotes à température ambiante) et il s’agit d’observer les microtubules qui contrôlent la réalisation de cette rotation corticale. 6.5 : Dans les zygotes contrôles, les microtubules sont alignés selon une direction préférentielle alors que dans les zygotes sans Trim36 on ne voit pas clairement de microtubules bien organisés et si certains sont présents, ils ne sont pas alignés selon une direction préférentielle. Cela indique que la rotation corticale ne peut se réaliser normalement ce qui entraîne un défaut de transport de Dishevelled du côté dorsal et un défaut de localisation nucléaire de β-caténine. 6.6 : On pourrait injecter de la protéine Dishevelled du côté dorsal du l’embryon ou également de l’ARNm de la β-caténine du côté dorsal. Cette surexpression permet de compenser la forte dégradation de la β-caténine causée par GSK3 et de lui permettre de rentrer dans le noyau et d’activer la cascade d’évènements qui aboutit à la formation du centre de Nieuwkoop et de l’organisateur de Spemann.
7.1 : Le croissant gris se forme lors de la rotation corticale et il se trouve toujours du côté dorsal. Donc c’est grâce à la pigmentation qu’on peut reconnaître les différentes destinées régionales des cellules. 7.2 : L’embryon surexprimant GBP dorsalement semble donner un têtard normal. En revanche, l’embryon surexprimant GBP ventralement a un double axe dorsal avec une duplication des régions antérieures. Cela rappelle les injections ventrales d’ARNm de β-caténine, de siamois ou de chordine. On peut imaginer que GBP est impliqué dans la même voie que ces protéines. 7.3 : L’irradiation aux UV désorganise le réseau de microtubules au pôle végétatif ce qui rend impossible la rotation corticale et donc la mise en place des axes de polarité. 7.4 : Comme prévu, l’irradiation aux UV provoque un indice DAI quasi-nul. De manière dose-dépendante, l’injection de l’ARNm de GBP permet de restaurer partiellement le phénotype normal (avec les doses utilisées, on n’arrive jamais à 5). Cela veut dire que GBP est suffisant pour compenser l’absence de rotation corticale. Il est probable qu’il se trouve en aval de la rotation corticale dans la cascade d’évènements aboutissant à la formation des axes de polarité. 7.5 : XGSK-3 s’oppose à l’action de GBP dans la compensation de l’absence de rotation corticale. XGSK-3 doit inhiber GBP ou alors la fonction de GBP est d’inhiber XGSK-3 mais l’injection d’ARNm de XGSK-3 rend impossible d’inhiber toute la quantité de XGSK-3 exprimée. Sachant que XGSK-3 est un inhibiteur de la β-caténine qui est indispensable pour la mise en place des axes, on peut imaginer que GBP fonctionne en inhibant XGSK-3 ce qui permet à la β-caténine de ne plus être dégradée et d’agir, ce qui peut parfaitement expliquer sa fonction de sauvetage des embryons sans rotation corticale. 7.6 : Dans l’hypothèse précédemment évoquée, GBP a une fonction similaire à Dishevelled.
8.1 : bc désigne le blastocoele et la pointe de flèche blanche indique le blastopore. 8.2 : On observe que le blastopore se forme même dans un explant sans ectoderme, et avec la même cinétique que sur un embryon entier ce qui montre que l’ectoderme n’est pas nécessaire pour la formation du blastopore.
9.1 : Alors que la calotte animale seule n’exprime pas d’actine musculaire (mais exprime bien l’isoforme de l’actine qu’on trouve dans tous types cellulaires), l’association avec les cellules de l’hémisphère végétatif provoque l’expression de l’actine musculaire. Les cellules de l’hémisphère végétatif sont des cellules endodermiques et donc on peut raisonnablement penser que ce sont les cellules de la calotte animale sous l’influence de signaux provenant des cellules endodermiques qui ont activé l’expression de l’actine musculaire car le muscle est un tissu mésodermique. Il s’agit ici de l’induction du mésoderme à partir de cellules ectodermiques sous l’influence de signaux provenant de l’endoderme. 9.2 : La déplétion du VegT d’origine maternel annule complètement l’activation de l’expression de l’actine musculaire dans les agrégats montrant que l’action de VegT est nécessaire pour l’induction du muscle (et sans doute de mésoderme, quelqu’il soit). 9.3 : Alors que la réexpression de VegT permet de sauver (au moins partiellement) l’activation de l’expression de l’actine musculaire (puits 7), l’expression de Bix4, pourtant un gène activé par VegT, ne suffit pas à sauver l’activation de l’expression de l’actine musculaire. On peut en déduire que Bix4 seul ne peut pas compenser l’absence de VegT et que sans doute d’autres cibles sont impliqués dans l’induction du mésoderme.
10.1 : Les embryons sans Wnt11 présentent une importante malformation. On ne distingue pas la formation claire de bourrelets neuraux alors qu’on est au stade neurula. Les embryons n’activent pas l’expression des cibles de la voie Wnt/β-caténine siamois et Xnr3, indiquant qu’aucun autre ligand Wnt n’a pu le remplacer dans cette fonction. L’injection des ARNm Wnt11 restaure plus ou moins bien le phénotype normal (plutôt bien pour l’embryon de gauche, moins bien pour celui de droite) et rétablit l’expression de siamois et de Xnr3. Cette expérience démontre que les ARN antisens ont bien eu une action spécifique (pas sur un autre gène Wnt par exemple) et qu’il n’ont pas induit de toxicité non lié à sa fonction. 10.2 : La surexpression de LRP6 (avec 75 pg d’ARNm) compense partiellement l’absence de Wnt11 en terme de morphologie (notamment sur l’embryon de droite où on observe des bourrelets neuraux). L’expression de siamois et de Xnr3 sont sauvés. Avec l’injection de 300 pg d’ARNm de LRP6, les embryons sont malformés et siamois et Xnr3 sont surexprimés, indiquant sans doute une hyperdorsalisation. 10.3 : La perte de LRP6 aboutit à une perte de la mise en place des axes de l’embryon et à une inhibition de l’expression de siamois et Xnr3. Ces défauts ne sont pas corrigés par la surexpression de Wnt11 montrant que Wnt11 a besoin de LRP6 pour pouvoir agir. 10.4 : Les expériences montrent que LRP6 est en aval de Wnt11 (si Wnt11 n’est pas là, LRP6 peut compenser mais l’inverse n’est pas vrai). Sachant que LRP6 est une protéine transmembranaire on peut émettre l’hypothèse que c’est un co-récepteur qui participe à l’activation de la voie Wnt/β-caténine.
11.1 : On observe une régression de l’axe antéro-postérieur de l’embryon et l’axe dorso-ventral semble lui aussi perturbé dans les situations C4 et C5. La structure céphalique y est particulièrement grosse ce qui fait penser à une dorsalisation/antériorisation. Le vitellus ne semble pas affecté. La surexpression de Bif1 a induit une mauvaise mise en place des axes de polarité. 11.2 : Comme prévu la surexpression de BMP2b provoque une ventralisation. La surexpression concomitante de Bif1 est capable de compenser plus ou moins totalement l’effet d’une quantité plus importante de BMP2b. Bif1 s’oppose aux effets des BMP, soit directement (en inhibant une des composantes de la voie), soit indirectement. Cela expliquerait pourquoi une surexpression de Bif1 aboutit à une dorsalisation puisque l’inhibition des BMP a cet effet. 11.3 : On observe une baisse de la phosphorylation de Smad1/5/8 alors que la quantité totale de ces protéines n’est pas modifiée. Bif1 est ainsi suffisante pour inhiber la voie de signalisation BMP et elle agit en amont de l’étape de la phosphorylation de Smad1/5/8. Cet effet explique ce qui a été observé dans l’expérience précédente.
12.1 : Sur les images, on observe que gsc est exprimé dans un côté du mésoderme (co-localisation avec une partie du marquage de ntla), le côté dorsal. D’après vos connaissances, vous devez savoir que gsc est exprimé dans l’organisateur de Spemann. 12.2 : La perte de fonction de foxh1 diminue la taille de l’organisateur de Spemann mais n’affecte pas de manière générale le mésoderme et l’endoderme. La perte-de-fonction d’eomesa perturbe fortement la formation de l’endoderme qui est réduit à sa portion dorsale. En ce qui concerne, le mésoderme (et notamment l’organisateur de Spemann), son étendue semble moins délimitée. 12.3 : Sans foxh1, ni eomesa, il n’y a pas d’endoderme et très peu de mésoderme avec disparition totale de l’organisateur de Spemann. En combinant ces résultats aux simples pertes-de-fonction, on en conclut que l’expression d’eomesa est nécessaire pour compenser l’absence de foxh1 lors de la formation du mésoderme (compensation cependant partielle pour l’organisateur de Spemann) et que l’expression de foxh1 est nécessaire pour compenser l’absence de eomesa lors de la formation de l’endoderme. 12.4 : Le gain-de-fonction de foxh1 étend l’organisateur de Spemann, démontrant que l’expression de foxh1 est suffisante pour cette fonction (dans une région précise néanmoins, donc d’autres facteurs doivent intervenir). Cette fonction est indépendante de l’expression d’eomesa car la perte de fonction d’eomesa ne perturbe pas cette extension. En comparant avec la perte-de-fonction d’eomesa seul, la surexpression de foxh1 fait plus que compenser le phénotype de réduction de l’organisateur de Spemann. Le gain-de-fonction de foxh1 ne semble pas affecter le développement de l’endoderme (peut-être un léger renforcement dorsal). La surexpression de foxh1 compense en partie la diminution de la taille de l’endoderme du mutant eomesa. On peut émettre l’hypothèse que foxh1 agit en partie en aval de eomesa ou alors agit via une voie indépendante sur les mêmes gènes cibles.
13.1 : Si le mésoderme est enlevé avant un certain stade, on remarque que l’endoderme ne forme pas de foie ou de poumons, ce qui indique que le mésoderme est nécessaire à l’induction de ces structures dans l’endoderme (et c’est le mésoderme qui, très probablement, envoie le signal inducteur). Au-delà de ce stade critique (25-26 pour le foie; 32-35 pour les poumons), les signaux inducteurs ont été envoyés et l’ablation du mésoderme n’a plus d’effets. 13.2 : La présence de mésoderme n’est pas aussi critique pour le développement du pancréas dans l’endoderme que pour le foie et les poumons aux stades étudiés. L’induction du pancréas a pu avoir lieu avant (et par exemple se terminer un peu après le stade 16, ce qui expliquerait l’expression plus faible des marqueurs de pancréas si on enlève le mésoderme à ce stade). La baisse de marqueurs de pancréas si on enlève le mésoderme au stade 32 est plus difficilement explicable. C’est peut-être un stade fragile/difficile à disséquer où l’ablation du mésoderme a pu endommager des cellules du pancréas.
14.1 : Les calottes animales isolées et laissées à se développer normalement donnent de l’épiderme cilié sous l’influence de la voie de signalisation BMP. Un dominant-négatif des récepteurs aux BMP bloque la voie de signalisation des BMP mimant l’action des molécules secrétées par l’organisateur de Spemann telles que Chordine qui induisent des tissus neuraux. 14.2 : Cela permet de suivre le devenir des différentes calottes, l’absence ou la présence de pigmentation étant utilisée comme un traceur de lignage. 14.3 : L’expression de Otx2 est fréquemment activée dans les calottes neuralisées indiquant que c’est du cerveau antérieur qui a été induit par le blocage de la voie BMP. Lorsque l’autre calotte exprime XMeis3, les calottes neuralisées n’expriment plus autant Otx2 et il s’agit d’un effet non cellulaire-autonome car les cellules qui expriment XMeis3 sont différentes de celles où l’expression de Otx2 est inhibée. On remarque aussi au passage que la partie neuralisée non pigmentée est alongée, indiquant un changement morphologique des tissus sous l’influence non cellulaire-autonome de XMeis3. 14.4 : Les calottes neuralisées n’expriment pas Krox20 en absence de XMeis3 dans la calotte voisine. Quand XMeis3 est exprimé, il induit de manière non-cellulaire autonome l’expression de Krox20 dans une bande étroite de cellules de la calotte neuralisée. On en déduit que XMeis3 a induit la postériorisation au moins partielle du tissu neural.
15.1 : Les explants au stade 16 expriment bien Snail2 tandis que les explants cultivés sans rien jusqu’au stade 22 n’expriment plus Snail2. Ces explants ne sont donc pas capables de maintenir la détermination de leurs cellules en crêtes neurales (ou alors ces cellules peuvent mourir). 15.2 : En co-culture avec du mésoderme intermédiaire, l’expression de Snail2 est maintenue au stade 22. Ce mésoderme doit donc envoyer un signal qui maintient les cellules de crêtes neurales dans les explants. 15.3 : En absence de mésoderme intermédiaire, l’ajout d’Edn1 dans le milieu suffit à maintenir l’expression de Snail2. En présence de mésoderme intermédiaire, l’inhibition de la signalisation endothéline inhibe le maintien de l’expression de Snail2 dans la majorité des explants, montrant que la signalisation endothéline est généralement nécessaire dans ce cas. On en déduit que l’action de l’Edn1 produite par le mésoderme intermédiaire essentielle pour maintenir la détermination (ou la survie) des cellules de crêtes neurales exprimant EdnrA dans le feuillet voisin.
16.1 Après la fécondation a lieu la rotation corticale qui laisse une région appelée « croissant gris » au futur côté dorsal de l’embryon. Le futur côté ventral est le côté opposé. 16.2 Chordine est exprimé dans l’organisateur de Spemann, c’est-à-dire dans le mésoderme dorsal. On observe sur les embryons non injectés que Wnt8 est exprimé dans la zone marginale de l’autre côté donc on en déduit qu’il est exprimé dans le mésoderme ventral. 16.3 Chez les embryons injectés avec l’ARNm codant Dscr6, on observe une expression ectopique de chordine dans du mésoderme plus ventral que l’organisateur de Spemann. En parallèle, une partie des régions ventrales n’exprime plus wnt8. Dscr6 favoriserait donc l’expression de chordine au détriment de wnt8. On peut interpréter ce patron d’expression comme le développement d’un deuxième centre organisateur de Spemann plus ventral. Si on les laissait se développer, les embryons devraient avoir deux axes dorsaux (tout comme les greffes classiques de Spemann-Mangold). 16.4 L’injection concomitante de l’ARNm de Stat3C avec l’ARNm de Dscr6 rétablit le phénotype normal (sauvetage) montrant que Stat3C s’oppose aux effets de Dscr6.
17.1 : Le domaine d’expression un peu faible de ephA4a qui correspond au rhombomère 1 s’étend postérieurement tandis que le marquage du rhombomère 3 disparait. En revanche, le marquage du rhombomère 5 flanqué par les vésicules otiques est seulement légèrement réduit. Le marquage de fgfr3 est étendu postérieurement jusque dans la région normalement marquée par krox20 au niveau du rhombomère 3 qui disparait. Le marquage du rhombomère 5 reste présent mais est réduit en taille. On en déduit qu’il y a une antériorisation des rhombomères. Notamment le rhombomère 3 (et probablement le rhombomère 2) deviennent indentiques au rhomobomère 1. 17.2 : Les perturbations de régionalisation selon l’axe antéro-postérieur sont accentués avec maintenant l’expression faible de ephA4a qui s’étend jusqu’au niveau de l’expression de hoxb4. La bande d’expression forte du rhombomère 5 a disparu, tout comme disparait l’expression de krox20. On en déduit que l’antériorisation des rhombomères est maintenant plus étendue et atteint même le rhombomère 5. 17.3 : On sait que Lzr/Pbx4 et Pbx2 sont nécessaires à l’action des facteurs de transcription Hox. Leur absence provoque une antériorisation des rhombomères et donc les facteurs Hox sont nécessaires pour donner une identité postérieure aux rhombomères plutôt que l’identité « par défaut » du rhombomère 1.