Par Patrick Pla, Université Paris-Saclay
Chez les organismes adultes, les tissus sont maintenus par un équilibre entre mort cellulaire et régénération. Cette homéostasie cellulaire est contrôlée par un ensemble de régulations croisées complexes entre les composants du cycle cellulaire, les molécules de signalisation et la matrice extracellulaire. Il arrive parfois que des cellules échappent à ces contrôles et se mettent à proliférer de manière incontrôlée. Ce dysfonctionnement se met en place progressivement, par accumulation de mutations et sélection des cellules les plus efficaces à échapper aux contrôles de leur prolifération.

- Un contrôle de l’intégrité du génome déficient
- Une résistance à la mort cellulaire
- Un cycle cellulaire hors de contrôle
- Des capacités migratoires exacerbées
- Un métabolisme particulier
- La capacité à faire former de nouveaux vaisseaux sanguins
- La capacité de bloquer les processus immunitaires anti-tumoraux
Un contrôle de l’intégrité du génome déficient
L’ADN génomique est soumis à des modifications d’origine interne (dépurinations, actions des espèces réactives de l’oxygène générées par le métabolisme…) et externe (radiations, molécules génotoxiques telles que le benzopyrène qu’on trouve dans la fumée du tabac,…). Les mécanismes de réparation de l’ADN sont dans ce contexte essentiels au maintien de l’intégrité des cellules. Cependant, les cellules tumorales sont issues de l’accumulation de mutations. Les enzymes de réparation de l’ADN y sont souvent défectueuses et en parallèle les mécanismes qui provoquent habituellement l’apoptose des cellules où trop d’anomalies génétiques se sont accumulées sont inhibés.
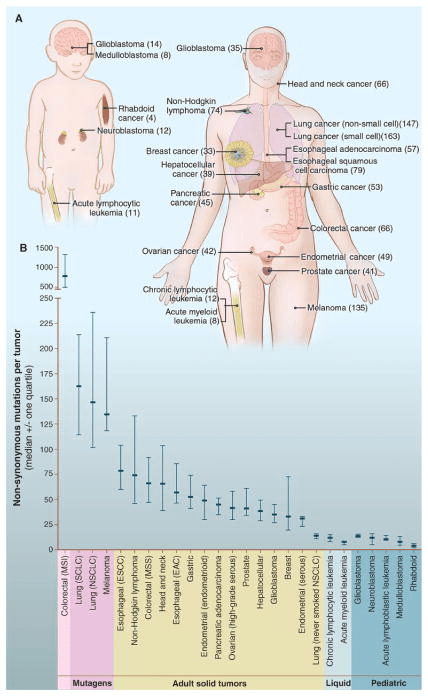
Des mutations ponctuelles peuvent s’accumuler mais des modifications de plus grande ampleur peuvent aussi être observées. Les fusions de gènes, résultant de réarrangements chromosomiques, sont des facteurs causatifs importants dans un large éventail de cancers. Ils représentent des indicateurs de première ligne pour le diagnostic, le pronostic et les biomarqueurs de sous-types de cancer, et servent de cibles pour le développement de médicaments.
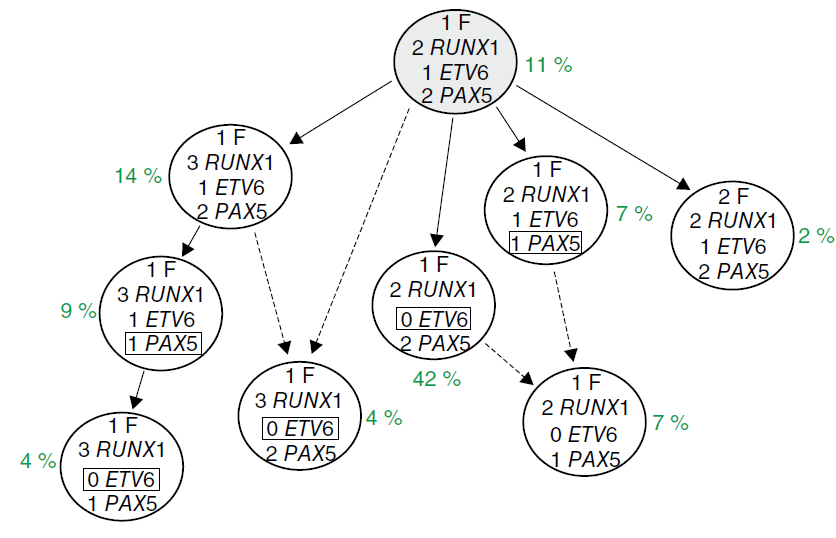
la lésion génétique initiale. Le nombre précédant chaque gène indique le nombre de copies du gène (sans compter celles du gène de fusion) et reflète donc les amplifications ou les délétions. On remarque que la délétion de PAX5 survient indépendamment dans deux sous-clones distincts. Les pourcentages représentent la représentation du profil génétique donné parmi les cellules tumorales. Source : https://www.academia.edu/download/45527132/Genetic_variegation_of_clonal_architectu20160510-8073-9c6vi6.pdf
Actuellement, plus de 25.000 fusions ont été identifiées dans 33 types de cancer qui correspondent à 16,5 % de cas de cancer (Gao et al., 2018). Dans les tumeurs d’origine hématologiques, on peut citer les fusions BCR-ABL dans la leucémie myéloïde chronique, PML-RARA dans la leucémie promyélocytaire aiguë, RUNX1-RUNX1T1 dans la leucémie myéloïde aiguë et SIL-TAL1 dans la leucémie aiguë lymphoblastique à cellules T pédiatrique.
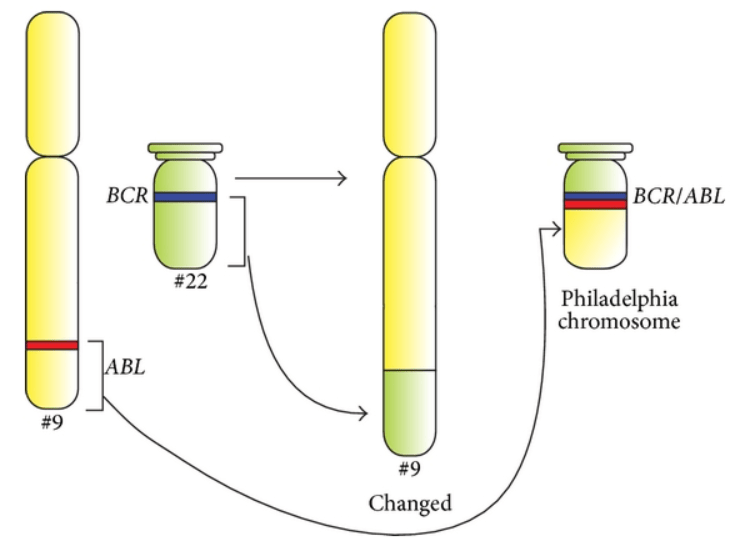
La fusion BCR-ABL est causée par une une translocation qui provoque échange de fragments d’ADN entre le chromosome 9 et le chromosome 22. Le chromosome 22 altéré possède alors les séquences codantes de deux gènes (BCR et ABL) côte à côte alors que normalement elles sont bien séparées (ABL sur le chromosome 9 et BCL sur le chromosome 22).
Vidéo sur pourquoi la protéine fusion BCR-ABL qui résulte de cette translocation est oncogène :
Une autre translocation « classique » est la translocation t(11;22)(q24;q12) menant à la formation de la fusion EWSR1:FLI1. Cela provoque le sarcome d’Ewing qui est une tumeur maligne des os et des tissus mous qui survient chez les enfants, les adolescents et les jeunes adultes. La protéine-fusion ESWR1:FLI1 se fixe anormalement sur des sites sur l’ADN qui activent l’expression de gènes dont les produits dérégulent le cycle cellulaire et la migration (Riggi et al., 2014).
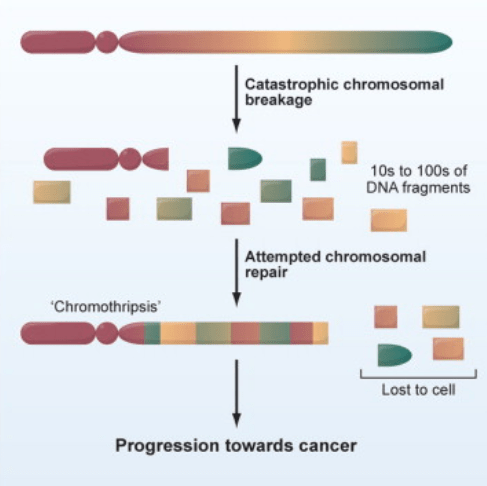
Les mutations génétiques peuvent s’accumuler graduellement mais aussi de multiples mutations peuvent avoir lieu simultanément comme lors de la chromothripsis qui est caractérisée par de multiples cassures double brin dans une région chromosomique. Ces fragments de chromosomes sont ensuite « racolés » ensemble par les protéines de réparation de l’ADN mais pas forcément dans le bon ordre. Cela concerne 2-3% des tumeurs (et 25% des tumeurs osseuses) (Stephens et al., 2011).
L’accumulation d’anomalies génétiques à différentes échelles que nous avons vu dans les exemples précédents provient de défauts dans les systèmes de réparation de l’ADN. Normalement, un dommage à l’ADN (cassure simple ou double brin, dimère de thymine induit par les UV…) est détecté et une voie de signalisation est activée provoquant l’intervention d’enzymes de réparation. Par exemple pour les cassures double brins :
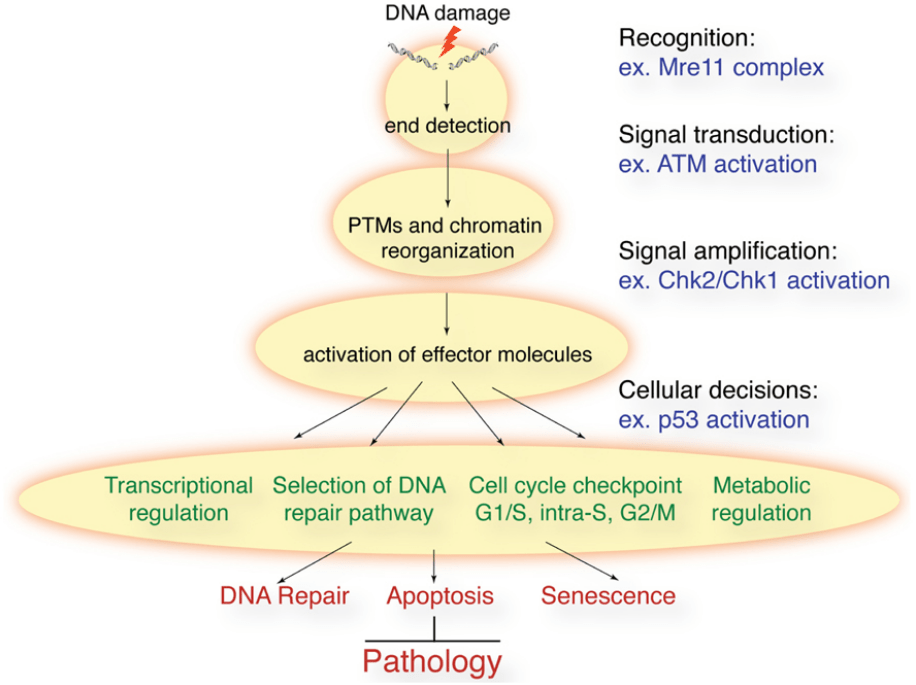
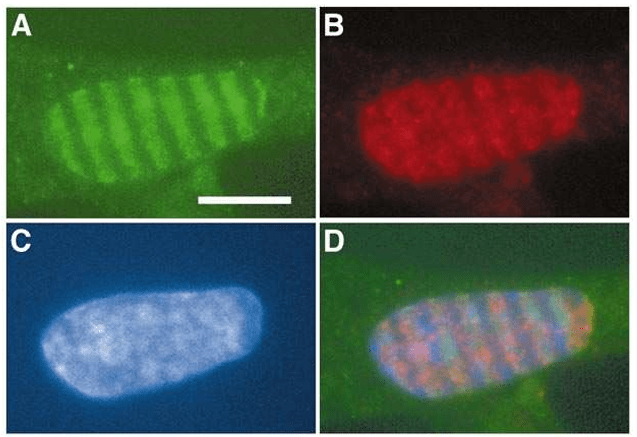
ATM contrôle les réponses aux dommages de l’ADN lors de la formation d’une cassure double brin de l’ADN. Il phosphoryle les histones de type H2AX sur la sérine 139 pour former γ-H2AX. Cette forme d’histone permet d’obtenir de l’ADN moins condensé ce qui facilite le recrutement des enzymes de réparation (Paull et al., 2000). La perte d’activité de la kinase ATM ou l’hétérozygotie pour les variants pathogènes du gène ATM augmentent les risques de cancer (Choi et al., 2016). La forme γ-H2AX n’est pas forcément précisément localisé strictement au point de la cassure double brin mais peut être présente jusqu’à plusieurs mégabases autour. Il y a ainsi une amplification du signal d’alerte.
γ-H2AX est reconnu par MDC1 qui déclenche ensuite le recrutement des enzymes de réparation (Lou et al., 2006).
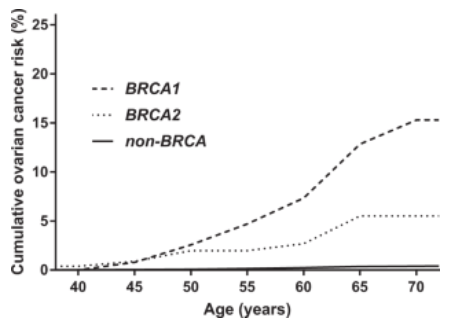
Parmi les protéines impliquées dans la réparation de l’ADN, il faut mentionner BRCA1 et BRCA2 dont des mutations des gènes sont impliquées dans 50% des cancers du sein et dans 75% des cancers de l’ovaire d’origine familiale.
Ils participent aux recrutements des nucléases (telle que Mre11) et des recombinases (tel que Rad51) qui interviennent lors de la réparation de l’ADN.
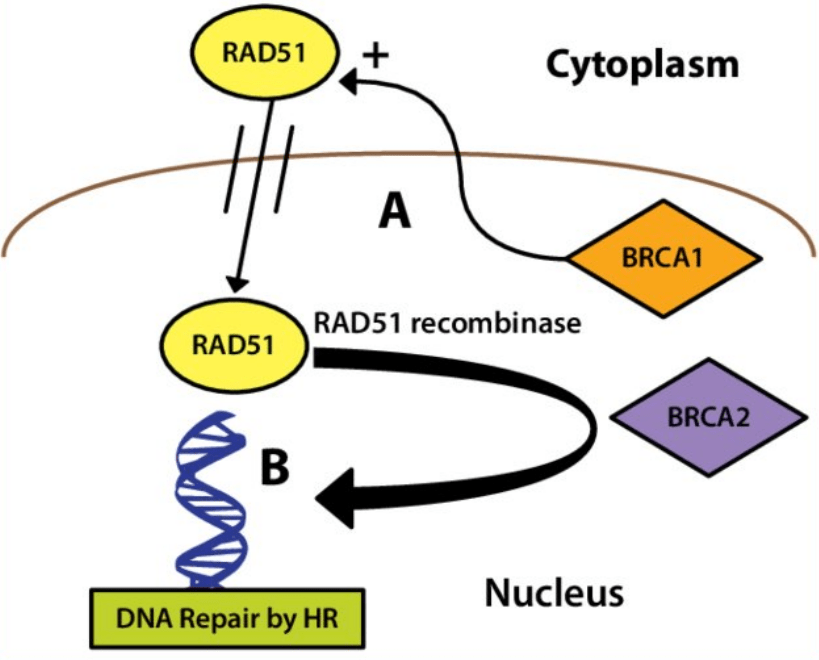
Source : https://www.researchgate.net/publication/221754538_The_relevance_of_BRCA_genetics_to_prostate_cancer_pathogenesis_and_treatment
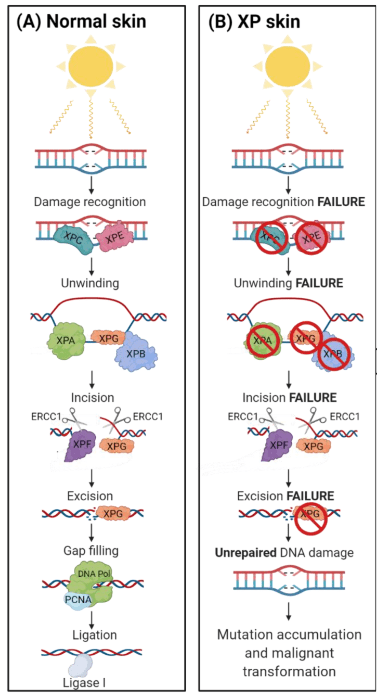
Xeroderma Pigmentosum (XP) est une maladie héréditaire rare (2,3/1 000 000 naissances en Europe occidentale) avec une pénétrance de 100 %, autosomique récessive, caractérisée par un défaut enzymatique dans la voie de réparation de l’ADN appelée réparation par excision de nucléotide (NER).
Les patients XP présentent généralement une sensibilité extrême à l’exposition aux UV (avec pour conséquence des coups de soleil douloureux), une sécheresse cutanée (également appelée xérose), des anomalies pigmentaires progressives ressemblant à des taches de rousseur (comme le suggère le terme « pigmentosum ») et une incidence accrue de tumeurs malignes de la peau au niveau du visage et du cou (Piccione et al., 2021).
Pour laisser le temps à la cellule de réparer son ADN, le cycle cellulaire doit être stoppé avant la réplication. C’est ce que réalise p53, un facteur de transcription. Habituellement, il est dégradé après ubiquitinylation à la suite de son association avec MDM2. MDM2 provoque aussi une inactivation des capacités de p53 à activer la transcription et stimule son export du noyau.
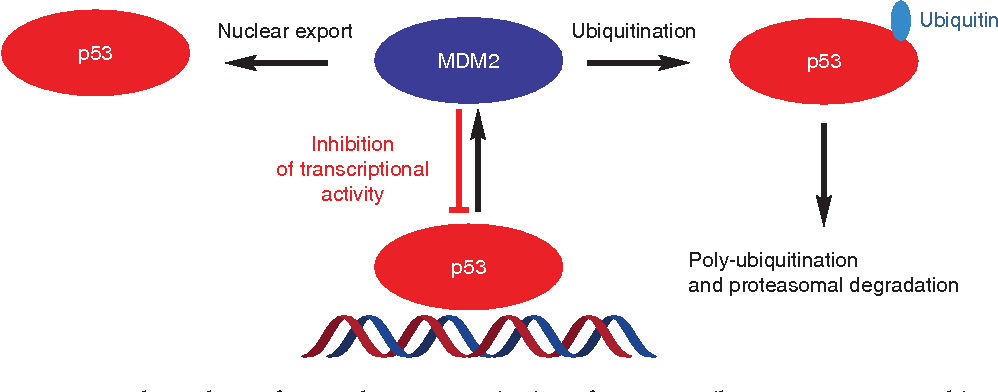
Lorsque des lésions endommagent l’ADN, les voies de signalisation activées inhibent MDM2 permettant à p53 d’agir en stoppant le cycle cellulaire. Si p53 n’est pas fonctionnelle (comme c’est le cas dans 50% des tumeurs), les cellules endommagées continuent de se répliquer, propageant aux cellules-filles des mutations non réparées.
L’instabilité du génome peut aussi provenir de dysfonctionnements de la mitose si les chromosomes ne sont pas répartis correctement entre les deux cellules-filles ce qui peut mener à une aneuploïdie : un nombre anormal de chromosomes dans les cellules. Cela peut être la conséquence par exemple de défauts dans la duplication du centrosome pendant la phase S menant à des fuseaux mitotiques multipolaires.
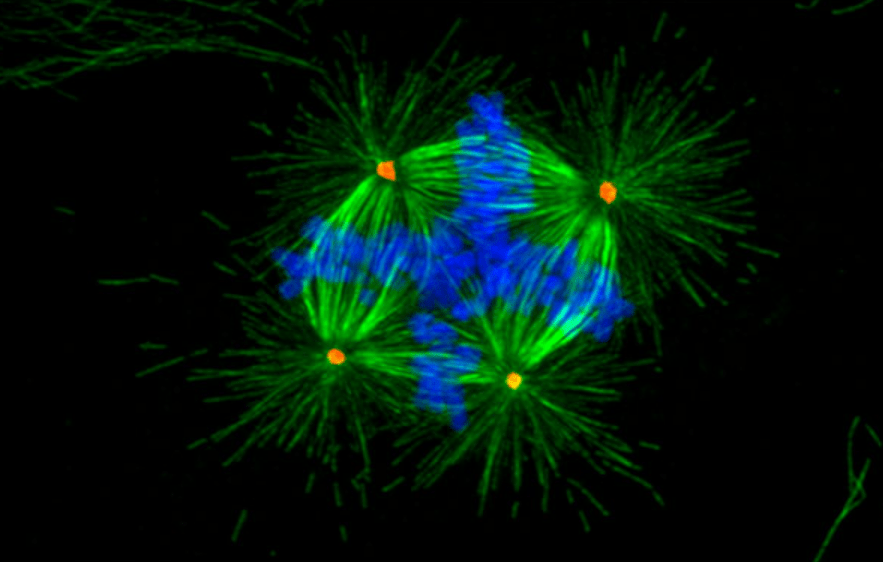
Des niveaux élevés d’aneuploïdie sont associés à un mauvais pronostic, notamment par une réponse immunitaire antitumorale réduite (Davoli et al., 2017). De plus, les chromosomes présentant davantage d’oncogènes et moins de gènes suppresseurs de tumeurs sont plus fréquemment amplifiés dans les cancers, c’est-à-dire les chromosomes 7, 8, 12, 13 er 20, les chromosomes 12 et 13 étant les plus fréquemment amplifiés, sans doute la résultante d’une pression de sélection (les aneuploïdies se font au hasard, mais certaines sont plus avantageuses que d’autres selon le contexte) (Davoli et al., 2013). Les tumeurs issues de différents tissus présentent des profils d’aneuploïdie spécifiques susceptibles de conférer aux cellules tumorales des avantages sélectifs particuliers à chaque type cellulaire (Sack et al., 2018).
Une résistance à la mort cellulaire
Les cellules meurent normalement par apoptose si elles ne reçoivent pas des signaux de survie de la part de leur environnement (ligands solubles ou composants de la matrice extracellulaire). L’apoptose est aussi déclenchée si les cellules accumulent trop de mutations qui ne peuvent pas être réparées.
Voir la page présentant l’apoptose de manière générale.
Les cellules tumorales arrivent à échapper à ces mécanismes d’activation de l’apoptose, souvent par perte de protéines promotrices d’apoptose (qui sont alors considérées comme codées par des gènes suppresseurs de tumeurs). Les cellules tumorales expriment aussi généralement des niveaux élevés de protéines anti-apoptotiques telles BCL-2, BCL-xL ou Mcl-1. Ces propriétés sont une des causes de la résistance des cellules tumorales aux traitements (Lin et al., 2017).
Des molécules inhibant BCL-2, BCL-xL ou Mcl-1 sont en cours de tests cliniques pour traiter des cellules tumorales rebelles aux traitements habituels (par exemple le navitoclax (Zhu et al., 2015)).
Ces molécules ont aussi pour effet de déclencher la mort de cellules sénescentes qui peuvent favoriser le développement de tumeurs à partir des cellules voisines.
A la suite de dommages à l’ADN qui ne sont pas réparées suffisamment rapidement, p53 active normalement l’apoptose. Elle réalise cette fonction notamment en stimulant la transcription du gène codant la protéine PUMA qui inhibe l’activité des acteurs anti-apoptotiques et stimule l’activité des acteurs pro-apoptotiques (Wang et al., 2007).
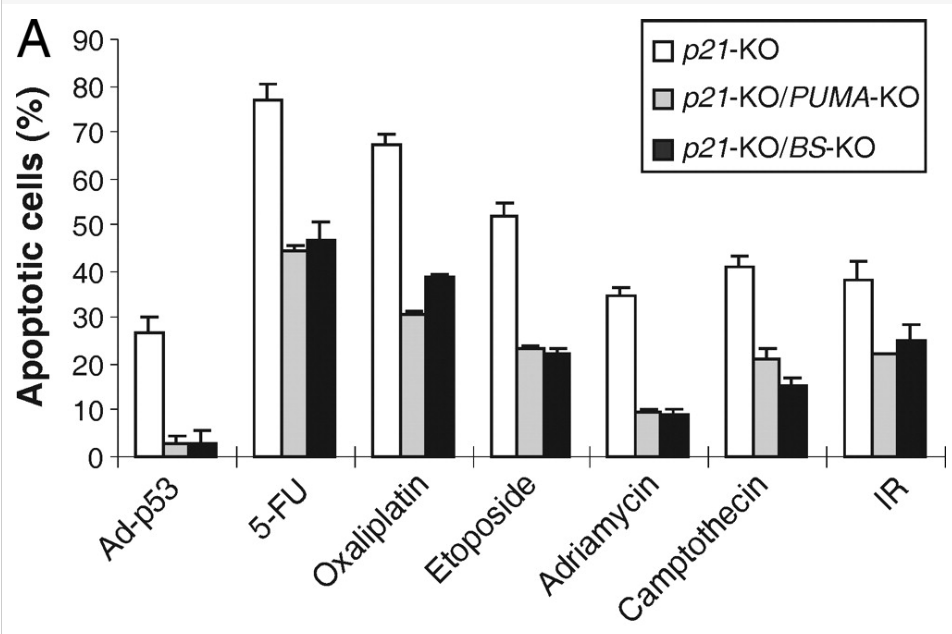
p53 est inactivé dans environ 50% des cancers, ce qui veut dire que dans ce cas, les mutations non réparées peuvent s’accumuler dans les cellules tumorales sans que l’apoptose ne soit déclenchée. Signalons que dans les cancers du col de l’utérus, c’est la protéine virale du papillomavirus HPV appellée E6 qui se fixe spécifiquement sur p53 et le fait dégrader par recrutement d’une ubiquitine ligase. Dans d’autres types de tumeurs, c’est un microARN, miR-3151 qui inhibe la production de p53 (Lankenau et al., 2015). L’inhibition de l’action de ce microARN augmente l’apoptose des cellules tumorales.
En plus de l’apoptose, signalons que la ferroptose est aussi parfois inhibée dans les cellules tumorales. La ferroptose est une mort cellulaire par accumulation de peroxides de phospholipides. Cette accumulation peut être provoquée par des ions Fe2+ d’où son nom. L’activation oncogénique de la voie PI3K-Akt-mTOR peut aboutir à une inhibition de la ferroptose par activation de l’expression de la stéaroyl-CoA desaturase-1 (SCD1), une enzyme du métabolisme lipidique qui a un effet protecteur (Yi et al., 2020).
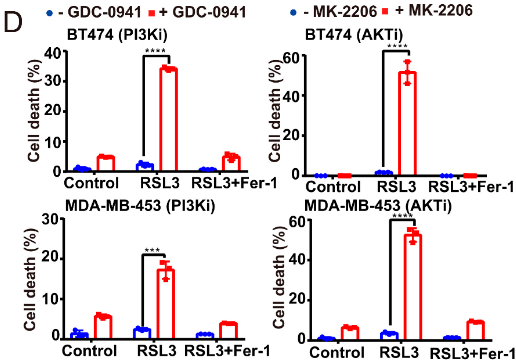
Un cycle cellulaire hors de contrôle
Revoir les notions de base sur le cycle cellulaire
Normalement, les cellules doivent recevoir un signal pour avancer dans le cycle cellulaire, répliquer leur ADN et entrer en mitose. Ce signal correspond souvent à la réception d’un ou de plusieurs facteurs de croissance sur des récepteurs qui activent alors des voies de signalisation intracellulaires. Ces voies permettent souvent de passer le point de restriction en G1 à partir duquel des facteurs de croissance ne sont plus nécessaires pour continuer dans les phases suivantes du cycle. Les cellules tumorales ont en général perdu la nécessité de recevoir ce signal, y compris en début de phase G1 où elles sont capables de passer le point de restriction sans l’aide de facteurs de croissance. Cela leur confère un avantage décisif par rapport aux cellules « normales » voisines.
Les protéines qui contrôlent directement le cycle cellulaire sont bien sûr au premier rang des altérations que l’on retrouve dans les cellules tumorales. Par exemple, le gène codant Rb présente deux allèles défectueux dans les tumeurs de l’oeil appelées rétinoblastome. La fonction normale de Rb est d’inhiber la progression du cycle cellulaire de G1 vers S. Souvent, les patients ont hérité de leur parent d’un allèle défectueux et lorsque l’autre allèle est muté dans certaines cellules, celles-ci ont beaucoup de chance de devenir tumorales.
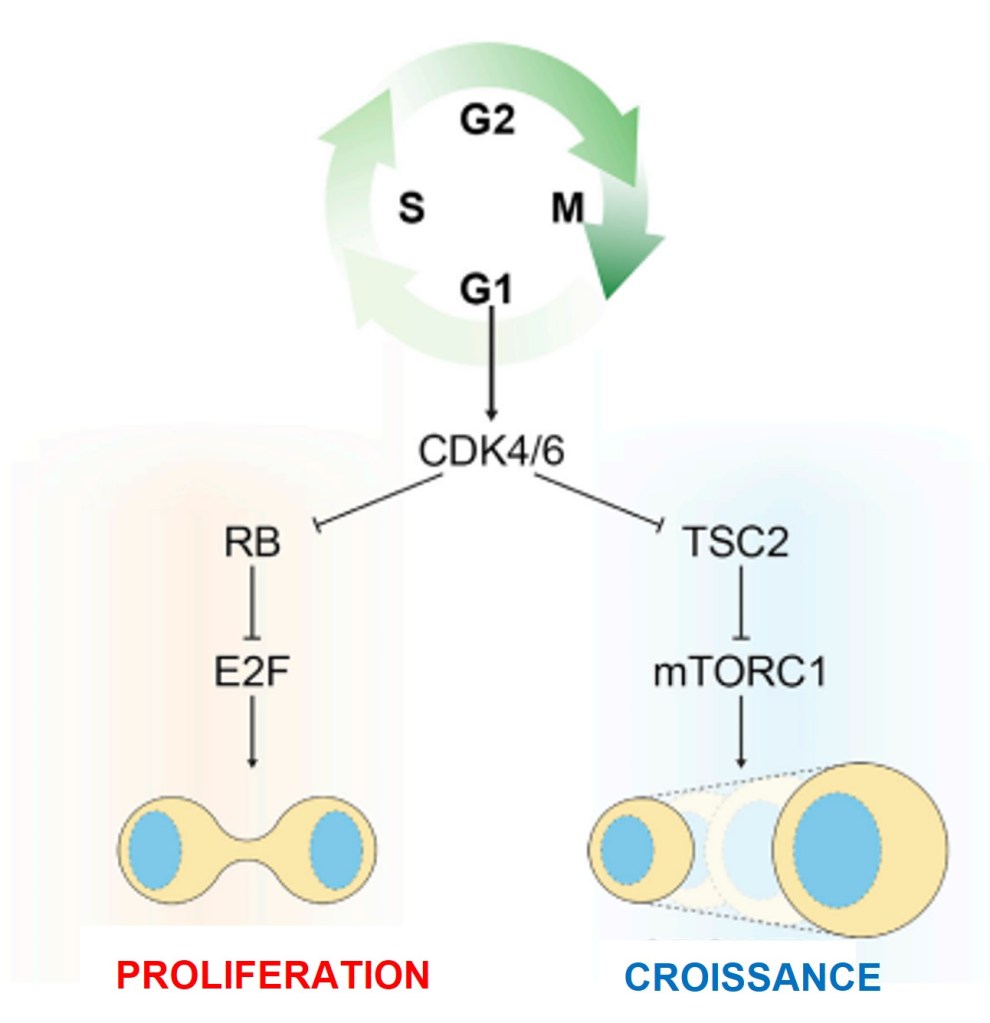
Rb est considéré comme un gène suppresseur de tumeur et comme on le voit sur cet exemple, les deux allèles doivent être atteints par une mutation perte-de-fonction pour favoriser le développement des tumeurs (propriété d’allèle récessif). C’est l’inverse pour les proto-oncogènes qui sont des protéines qui favorisent l’avancée dans le cycle cellulaire et où un seul allèle gain-de-fonction (qui transforme ce proto-oncogène en oncogène) suffit à favoriser la tumorigenèse (propriété d’allèle dominant).
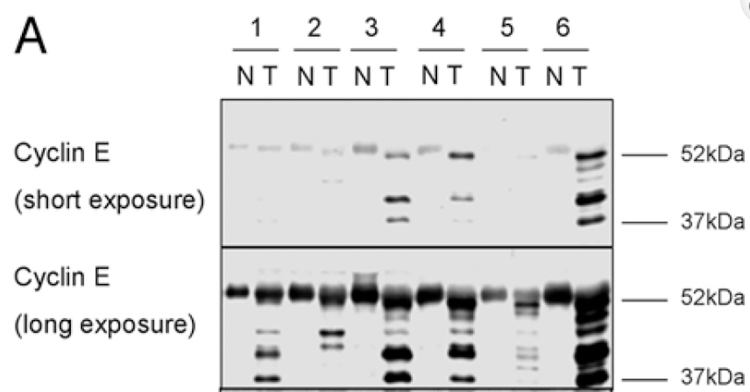
Bien sûr des dérégulations des cyclines sont impliquées dans la formation de tumeurs. Par exemple, des isoformes de la cycline E de faible poids moléculaire exprimées dans certaines formes de cancer du sein sont résistantes aux CKI (inhibiteurs de kinase-cycline dépendantes), se lient plus efficacement à CDK2 et peuvent ainsi stimuler plus efficacement la progression du cycle cellulaire (Wingate et al., 2009). Ce sont des marqueurs de pronostic du cancer du sein à un stade précoce (avec ganglions négatifs) (Sutherland et al., 2002).
Des récepteurs à des facteurs de croissance peuvent être mutées dans les cellules tumorales et être constitutivement actives, c’est-à-dire capables d’activer les voies de signalisation en aval, même en l’absence de ligand. Les récepteurs peuvent aussi être surexprimés par mutations dans les séquences régulatrices de leur gène ou par amplification génique. C’est le cas par exemple du récepteur aux EGF (EGFR) dans certaines tumeurs du poumon ou du cerveau.
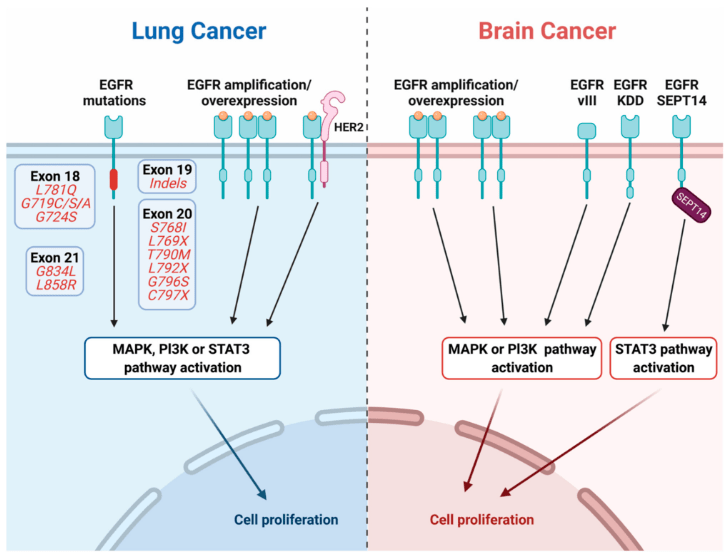
HER2/ErbB2 qui est aussi un récepteur aux EGF est surexprimé dans 25% des cancers du sein (Slamon et Clark, 1988). Dans 50% des cas des cancers du sein triplement négatif (pas d’expression de HER2/Erb2 et de récepteurs aux oestrogènes et à la progestérone), il y a une surexpression de EGFR (Alvarez et al., 2010).
En aval des récepteurs, le long des voies de signalisation, des mutations et des surexpressions des différents acteurs peuvent aussi intervenir dans les processus tumoraux. Par exemple, en aval des récepteurs de la famille des EGFR, la suractivation de STAT3 ou de STAT5 favorise la progression des cellules vers un stade tumoral. Par ailleurs, Cette voie peut aussi être activée par des cytokines pro-inflammatoires ce qui permet de lier inflammation et tumorigenèse (Johnson et al., 2018).
La mutation BRAFV600E se retrouve dans la moitié des cas de mélanomes. Le changement de la valine en acide glutamique change la conformation de la sérine/thréonine kinase BRAF et la rend constitutivement active alors que d’habitude, son activité nécessite la fixation d’un facteur de croissance sur un récepteur de la cellule. D’autres mutations sont aussi trouvées sur le même site comme BRAFV600K de manière moins fréquente mais avec des effets similaires.
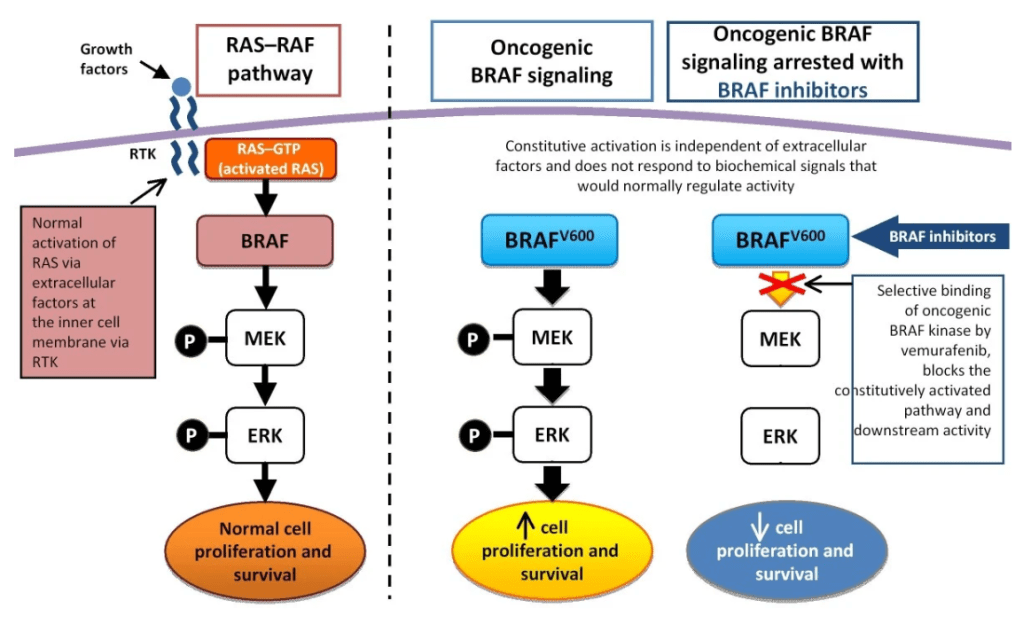
Une version commentée de cette figure est disponible en vidéo.
Source : https://translational-medicine.biomedcentral.com/articles/10.1186/1479-5876-10-85
La protéine SRC (prononcée « sarc », pour sarcome) est une tyrosine kinase non-réceptrice codée par le gène SRC. Elle a été initialement identifiée comme l’oncogène viral v-src dans le virus du sarcome de Rous (rétrovirus qui cause des tumeurs chez le poulet et dont l’étude a permis de découvrir les oncogènes). Sa version cellulaire, c-SRC, est la premier proto-oncogène découverte. SRC fait partie d’une famille de kinases impliquées dans des processus de signalisation intracellulaire essentiels au maintien de l’homéostasie cellulaire. L’activité de SRC est strictement contrôlée, notamment via la phosphorylation de la tyrosine 527 (Y527), qui maintient la protéine inactive. Sa déphosphorylation ou certaines mutations oncogéniques mettent la kinase dans une conformation active.
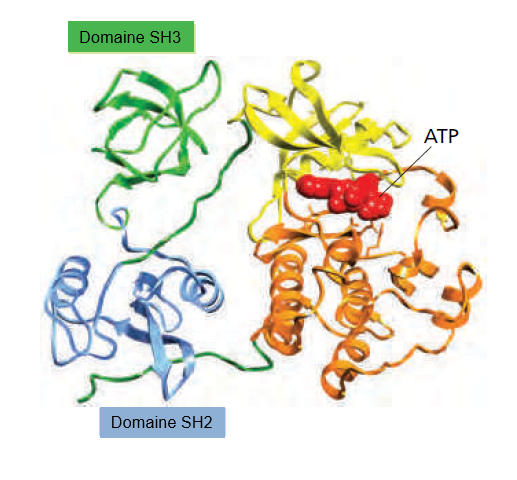
La protéine Src est une tyrosine kinase non réceptrice impliquée dans la signalisation cellulaire. Elle comporte plusieurs domaines fonctionnels :
Domaine SH3 (en vert) : reconnaît les motifs riches en prolines présents dans les protéines partenaires. Domaine SH2 (en bleu) : se lie spécifiquement aux résidus tyrosine phosphorylés. Domaine catalytique kinase (en jaune et orange) : responsable de l’activité enzymatique de Src ; il lie l’ATP (en rouge), nécessaire pour transférer un phosphate sur les tyrosines cibles. Ces domaines coopèrent pour réguler l’activité de Src, qui joue un rôle clé dans la prolifération, la survie et la migration cellulaire. Une activation anormale de Src peut contribuer au développement de cancers. D’après https://www.rcsb.org/structure/2SRC#:~:text=RCSB%20PDB%20%2D%202SRC%3A%20CRYSTAL%20STRUCTURE,IN%20COMPLEX%20WITH%20AMP%2DPNP
Dans de nombreuses tumeurs solides (côlon, sein, poumon, pancréas…), une augmentation de l’expression et/ou de l’activité de SRC est corrélée à un mauvais pronostic clinique et à des formes plus agressives de la maladie. L’hyperactivation de SRC stimule la division cellulaire et protège les cellules tumorales de l’apoptose, notamment via Ras/MAPK et PI3K/Akt (Chatzizacharias et al., 2012; Wheeler et al., 2012). Elle est impliquée dans la résistance des tumeurs à la chimiothérapie (Raji et al., 2024).
La protéine APC est codé par un gène suppresseur de tumeur. Elle fait partie d’un complexe qui favorise la phosphorylation de la β-caténine par GSK3 et sa dégradation. Si la β-caténine n’est pas dégradée, elle rentre dans le noyau et se lie aux facteurs de transcription LEF/TCF et active la transcription de gènes favorisant la prolifération comme c-Myc ou Cycline D. Une baisse d’expression ou une mutation perte-de-fonction d’APC provoque l’entrée systématique de la β-caténine dans le noyau et l’activation continuelle de la transcription de gènes favorables à la prolifération. On trouve fréquemment des mutations ou des surexpressions d’APC dans les cellules tumorales colo-rectales.

Le gène suppresseur de tumeur BRCA1 code une protéine qui participe aux voies de réparation des cassures double-brin de l’ADN mais qui interagit aussi directement avec le récepteur aux oestrogènes ER-α en inhibant son action qui favorise la prolifération.
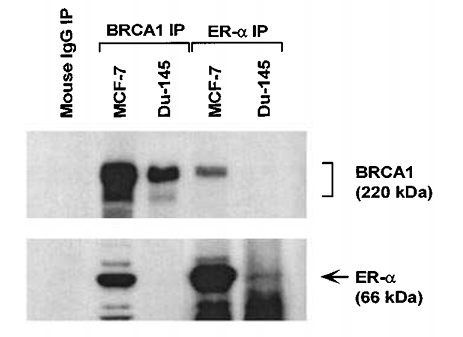
Les mutations perte-de-fonction de BRCA1 se retrouvent tout particulièrement dans les cellules tumorales qui répondent aux oestrogènes (dans les glandes mammaires et les ovaires notamment). Quand BRCA1 est moins présent, le récepteur ER-α est « trop » actif en présence d’oestrogènes et la prolifération est trop stimulée pour que les réparations à l’ADN puissent se faire correctement (Wang et al., 2014). Tout comme pour p53, on voit donc que réparation de l’ADN et avancée dans le cycle cellulaire sont liées et que l’équilibre entre ces deux processus est rompu dans les cellules tumorales.
Normalement, le nombre de divisions qu’une cellule puisse réaliser est limité par le raccourcissement des extrémités de ses chromosomes, appelés télomères.
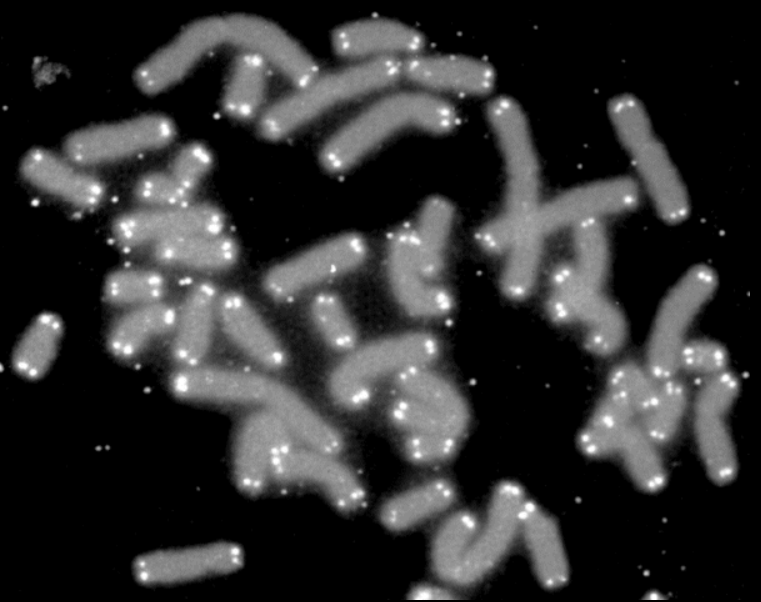
Les télomères sont constitués d’une répétition d’une séquence de 5 à 11 pb (TTAGGG chez les Vertébrés). Le raccourcissement progressif des télomères est observé dans les cellules en culture et in vivo, et constitue une caractéristique du vieillissement qui est souvent appelée « horloge des télomères » (Harley et al., 1992).
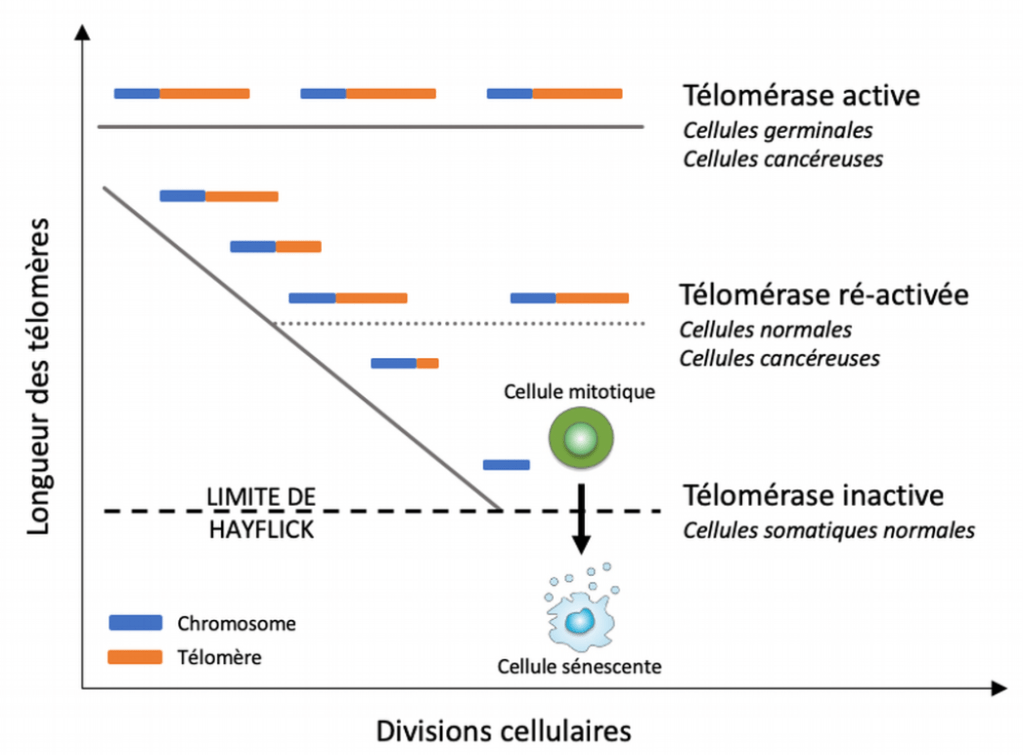
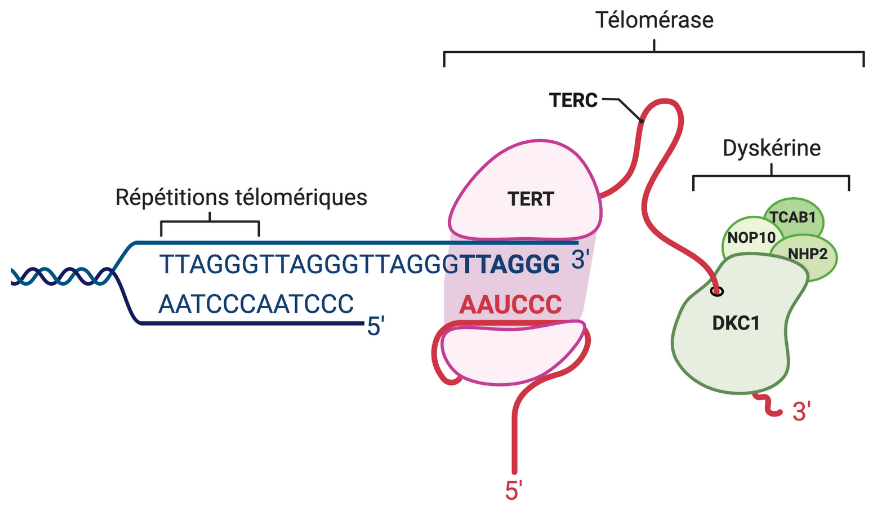
L’érosion des télomères est la conséquence des mécanismes de la réplication et du besoin d’une amorce pour l’ADN polymérase. Elle se produit dans les cellules qui manquent d’activité suffisante d’une enzyme appelée télomérase, dont le noyau catalytique est composé d’une transcriptase inverse (TERT) et d’un ARN appelé TERC qui compensent la perte des séquences télomériques dues à la réplication.
Sans télomérase, une baisse inexorable des séquences télomériques conduit finalement à ce que l’on appelle le décapage des télomères. Ce décapage provoque une réponse aux dommages à l’ADN qui entraîne une perte et/ou des réarrangements de matériel génétique et la mort (Lazzerini-Denchi et Sfeir, 2016; Muraki et al., 2012).
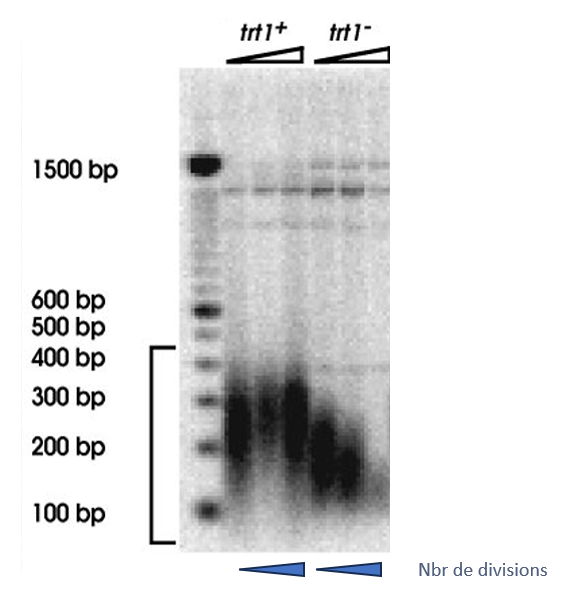
Néanmoins, les cellules tumorales échappent à cette limite car elles réexpriment très souvent la télomérase. C’est le cas par exemple dans 85% des cancers de l’estomac (Yakoob et al., 1999). Autre exemple, lors de l’établissement du pronostic du neuroblastome, une tumeur pédiatrique du système nerveux périphérique, le niveau d’activité élevé de la télomérase est associé à une mauvaise réponse au traitement et à une évolution plus défavorable (Yu et al., 2022). La voie ALT qui est un mécanisme d’allongement des télomères basé sur la recombinaison peut aussi être activée.
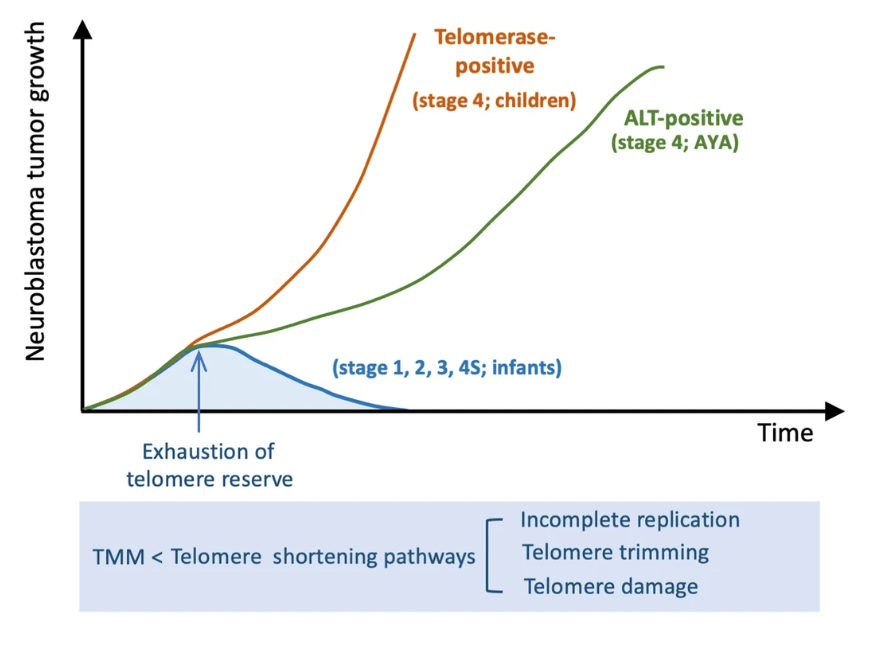
Le niveau d’expression de facteur de transcription N-myc, qui régule l’expression de la télomérase, est également prédictif de l’évolution de ces tumeurs. Les tumeurs ayant amplifié le gène N-myc avec un nombre de copies élevé ont le pire pronostic.
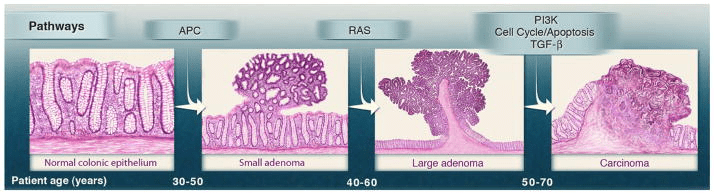
Il est fréquent que des tumeurs se forment à partir de cellules qui ont déjà tendance à proliférer, notamment les cellules souches adultes. Par exemple, une part substantielle des tumeurs du cancer du colon, provient des cellules souches Lgr5+ qui se trouvent au fond des cryptes et se divisent régulièrement (en situation normale) pour régénérer l’intestin sur une période de 5 jours. La mutation du gène suppresseur de tumeur codant APC (impliqué dans la modération de la voie Wnt) est fréquente dans les cancers du colon et les chercheurs ont montré que l’inactivation de ce gène dans les cellules souches Lgr5+ suffit à provoquer des tumeurs (Barker et al., 2008). Dans un autre tissu, la surexpression des membres de la famille des Wnt a montré qu’ils étaient des régulateurs importants du développement cellulaire normal, conduisant à l’expansion
du pool de cellules souches mammaires, mais que cela augmente la susceptibilité au cancer (Liu et al., 2004).
Une partie de l’arsenal de chimiothérapie anti-tumorale repose sur des molécules qui empêchent l’assemblage des microtubules (vincristine, vinblastine) ou, au contraire, empêche leur désassemblage (paclitaxel ou taxol) ce qui bloque le déroulement des mitoses. Malheureusement, les mitoses des cellules « normales » sont également affectées en même temps que celles des cellules tumorales ainsi que d’autres processus cellulaires impliquant les microtubules ce qui provoque de lourds effets secondaires.
Des capacités migratoires exacerbées
Revoir les notions de base sur la migration et la transition épithélio-mésenchymateuse
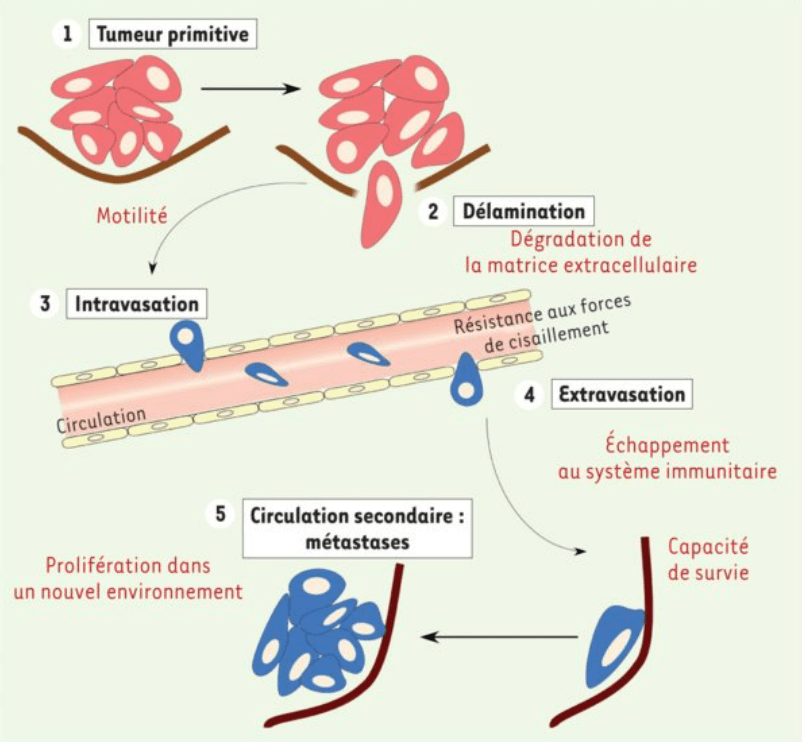
Les cellules tumorales à leur site primaire peuvent causer des dysfonctionnements mais 90% de la mortalité des patients atteints de cancers provient des métastases qui sont issues de la tumeur primaire par migration et transport lymphatique ou sanguin.
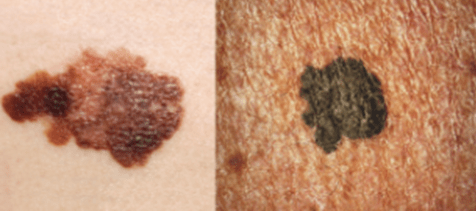
Les mélanocytes de la peau peuvent donner naissance à des mélanomes qui sont parmi les plus cellules tumorales les plus invasives lorsqu’elles forment des métastases (25% de survie à 5 ans chez les patients avec métastases contre 99% si le mélanome reste in situ). C’est parce qu’elles réactivent des réseaux génétiques proches de ceux des crêtes neurales.
La première étape de la formation des métastases pour les cellules tumorales d’origine épithéliales est constituée par la transition épithélio-mésenchymateuse (EMT). On retrouve dans les cellules tumorales de nombreuses voies de signalisation et de nombreux facteurs de transcription pour coordonner leur EMT qui sont commun aux EMT lors du développement embryonnaire, comme par exemple lors de la gastrulation des Amniotes ou lors du développement des cellules de crête neurale.
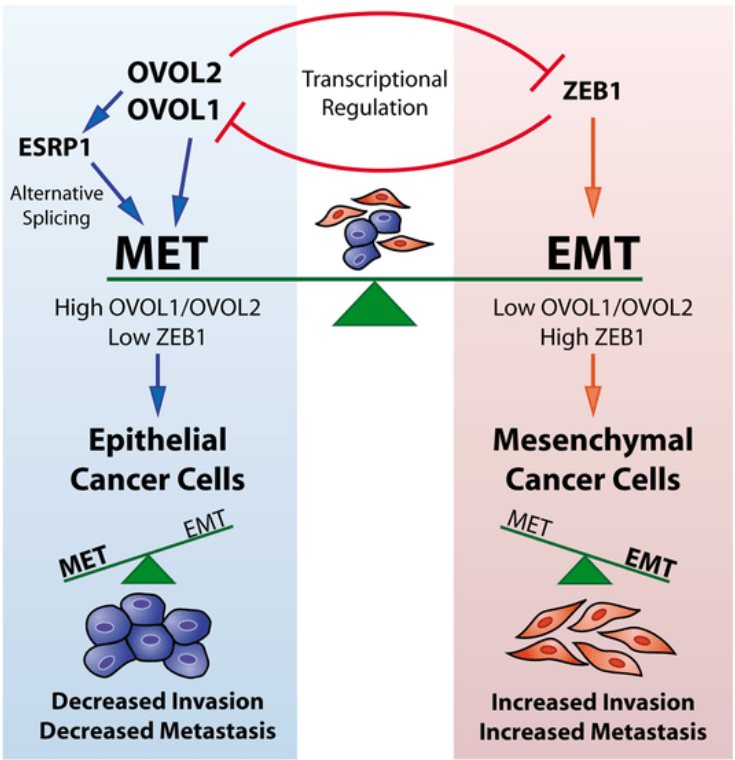
Dans les cellules cancéreuses humaines, les états mésenchymateux et épithéliaux sont induits et maintenus par des programmes de régulation transcriptionnels et post-transcriptionnels. Ces programmes sont contrôlés par la régulation par rétroaction entre les facteurs de transcription OVOL et ZEB1, inducteurs critiques respectivement de MET (transition mésenchymato-épithéliale) et EMT. De plus, ces facteurs de transcription contrôlent l’expression d’ESRP1, un régulateur de l’épissage crucial pour le MET et réprimé dans l’EMT. Par conséquent, un niveau d’OVOL élevé et un niveau de ZEB1 faible stabilisent l’état épithélial en diminuant l’invasion des cellules cancéreuses et les métastases, et inversement pour l’état mésenchymateux. Source : https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0076773
Le facteur de transcription TWIST1 est exprimé dans de nombreux types de cancers (Yang et al, 2004), et son expression est associée à de mauvais pronostics pour les patients (Banerjee et al, 2011; Riaz et al, 2012).
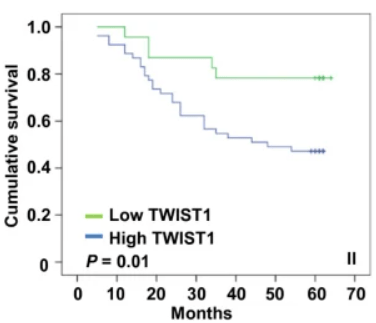
TWIST1 induit l’EMT et augmente la mobilité de nombreux types de cellules cancéreuses (Yang et al, 2004 ; Mani et al, 2008 ; Qin et al, 2012 ; Xu et al, 2017). Après l’EMT, TWIST1 peut augmenter la migration et l’invasion des cellules cancéreuses en stimulant la formation d’invadopodes et en accélérant le renouvellement du complexe d’ahérence focaux (Eckert et al, 2011 ; Yang et al, 2016).
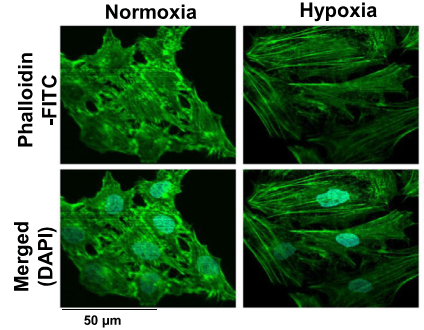
L’hypoxie qui est présente dans le coeur de nombreuses tumeurs est un facteur qui favorise l’EMT (Hapke et al, 2020).
Les cellules épithéliales qui deviennent métastatiques perdent leur polarité apico-basale. Le complexe Crumb est crucial pour maintenir cette polarité et des cellules tumorales invasives rénales ou mammaires présentent une expression anormalement faible de l’un des composants du complexe, CRB3 (Mao et al., 2015). Les cellules exprimant trop peu CRB3 perdent aussi l’inhibition de contact (Mao et al., 2018).
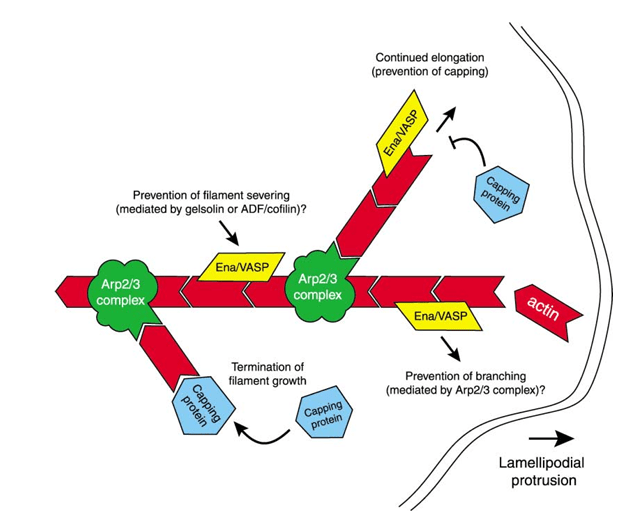
Très logiquement, les protéines contrôlant le réseau de microfilaments qui est responsable de la formation des lamellipodes et des invadopodes impliqués dans la migration ont un fonctionnement altéré dans les cellules métastatiques. C’est le cas par exemple des protéines de la famille Ena/VASP comme MENA dans les cancers du sein (Di Modugno et al., 2006, Klemke, 2012). Au stade avancé de la tumeur, cette famille des protéines est surexprimée ce qui augmente la vitesse de migration in vivo et stimule le caractère invasif des cellules.
La voie de signalisation canonique Wnt semble importante pour les capacités migratoires des cellules tumorales. Les cellules d’adénocarcinome pulmonaire métastatique isolées à partir de ganglions lymphatiques présentent une activité TCF/LEF augmentée, suggérant que les cellules où la voie Wnt est fortement activée sont responsables des étapes initiales de la métastase (Nguyen et al., 2009). Dans le glioblastome, les cellules à haut niveau d’activation de la voie Wnt peuvent s’attacher et envahir plus efficacement les tissus le long du système vasculaire (qui est le mode de propagation classique dans le glioblastome) (Griveau et al., 2018). Il a été montré que la suppression de l’expression de la β-caténine réduit la migration et l’invasion in vitro dans un modèle de carcinome épidermoïde de la tête et du cou (HNSCC) (Moon et al., 2021). Cependant des données récentes montrent que dans certains cancers, l’intravasation (l’entrée dans les vaisseaux sanguins), le transport dans le sang ou la lymphe et l’extravasation (sortie des vaisseaux sanguins) à l’origine des métastases s’accompagne d’une baisse transitoire de l’activité de la voie de signalisation Wnt canonique et d’une hausse de l’activité de la voie non canonique (voie PCP) (Stoletov et al., 2023).
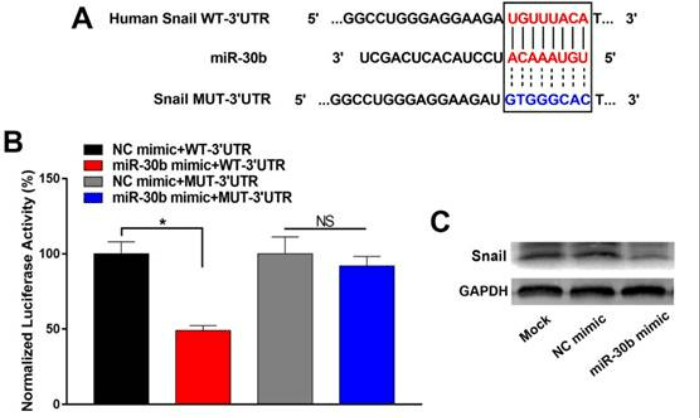
Des régulations au niveau traductionnel peuvent empêcher l’EMT et la migration des cellules tumorales notamment grâce à l’action du microARN miR-30b qui cible directement le principal facteur de transcription coordonnant l’EMT, Snail et empêche sa traduction. L’expression de ce microARN est diminuée dans les cellules métastasiques agressives (Zhang et al., 2020).
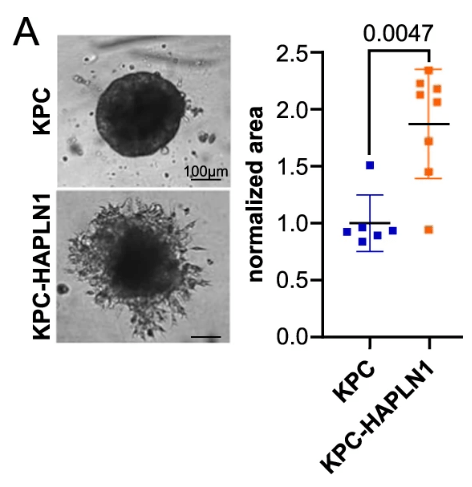
L’environnement des cellules tumorales joue également un rôle primordial dans la formation de métastases, en premier lieu la matrice extracellulaire (MEC). Par exemple, la présence de HAPLN1 (une protéine liée aux hyaluranes et aux protéoglycanes) facilite la formation de métastases péritonéales à partir de cellules tumorales du pancréas (Wiedmann et al., 2023).
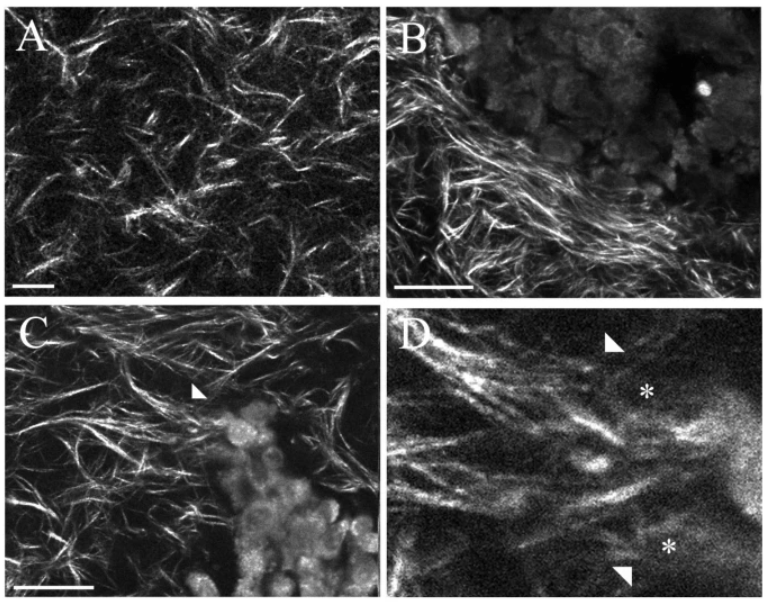
Les structures du collagène diffèrent selon l’emplacement dans la tumeur, et présentent des niveaux d’alignement plus élevés au bord de la tumeur, facilitant la migration cellulaire (Provenzano et al., 2006; Provenzano et al., 2008). Il semble que cet alignement des fibres de collagène soit provoqué par les cellules tumorales elles-mêmes.
Autre exemple où les cellules tumorales peuvent participer à créer un environnement propice à leur migration : des cellules faiblement métastatiques issues de cancer du sein secrètent des vésicules extracellulaires riches en Transglutaminase 2 (Tg2) qui activent les fibroblastes environnants à produire une matrice extracellulaire plus favorable à la migration (Schwager et al., 2022).
Parmi les éléments de l’environnement favorable aux cellules métastatiques figure le système immunitaire et notamment les macrophages qui sont impliqués dans une boucle paracrine avec les cellules tumorales au cours des stades métatastatiques initiaux. Le chimiotactisme des cellules métastatiques vers les vaisseaux sanguins se produit en réponse à des molécules telles que l’EGF sécrétés par les macrophages associés aux vaisseaux. Les cellules cancéreuses expriment le récepteur de l’EGF (EGFR) et sécrètent CSF-1, qui attire les macrophages et les incite à exprimer l’EGF, complétant ainsi une boucle de rétroaction paracrine positive. La signalisation entre le macrophage et la cellule tumorale affecte l’activité des régulateurs de l’actine tels que WASP et N-WASP, entraînant la formation de podosomes dans les macrophages et des invadopodes dans les cellules tumorales qui favorisent l’intravasation des cellules tumorales (Condeelis et Pollard, 2006).
La migration est souvent étudiée à partir de la tumeur primaire mais pour la formation des tumeurs secondaires, il ne faut pas oublier les étapes d’extravasation (sortie des vaisseaux sanguins vers le site de colonisation) et la migration à travers le nouveau tissu.
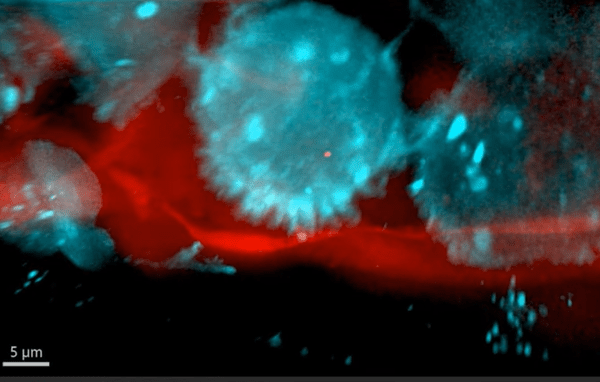
Signalons que des migrations collectives de cellules tumorales ont été observées. Dans ce cas, les cellules restent adhérentes entre elles. Au sein d’un amas tumoral en migration, une minorité de cellules se localisent en périphérie, dans le sens de la migration, et mènent le groupe. Ces cellules, appelées cellules leaders, présentent des forces de protrusion actives, interagissent avec la matrice extracellulaire (MEC) environnante et génèrent des forces de traction. Dans le cancer du sein, les cellules leaders sont définies histologiquement par l’expression de la kératine 14 (K14), une protéine des filaments intermédiaires (Cheung et al., 2016). Les fibroblastes associés au cancer (CAF) peuvent également être présents dans les amas tumoraux en migration et contribuer à l’invasion et à la migration collectives des tumeurs (Bayer et al., 2019).
Un métabolisme particulier
L’une des caractéristiques les plus fréquemment observées dans les tumeurs humaines est la surexpression et la suractivation des transporteurs de glucose à la surface des cellules cancéreuses (Macheda et al., 2005). Une surexpression de ces transporteurs peut être associée à une résistance à la chimiothérapie (Chang et al., 2021). Les besoins métaboliques de cellules tumorales sont très importants mais c’est non seulement quantitativement mais aussi qualitativement que leur métabolisme glucidique et énergétique est altéré.
En présence d’oxygène, les cellules métabolisent normalement le glucose en pyruvate via la glycolyse. Le pyruvate est alors transporté dans les mitochondries, où il est introduit dans le cycle de Krebs. Puis la chaîne respiratoire mitochondriale permet la production importante d’ATP via la phosphorylation oxydative. L’oxygène est alors nécessaire comme accepteur final d’électrons. Si l’oxygène est en quantité limitante (anoxie), les cellules métabolisent le pyruvate en lactate, une réaction catalysée par la lactate déshydrogénase, permettant à la glycolyse de se poursuivre en recyclant le NADH en NAD+. Les cellules cancéreuses et les cellules en prolifération ont un métabolisme particulier convertissent la plupart du glucose en lactate, que l’oxygène soit présent ou non. Il s’agit de l’effet Warburg (du nom d’Otto Warburg qui a découvert ce phénomène en 1924).
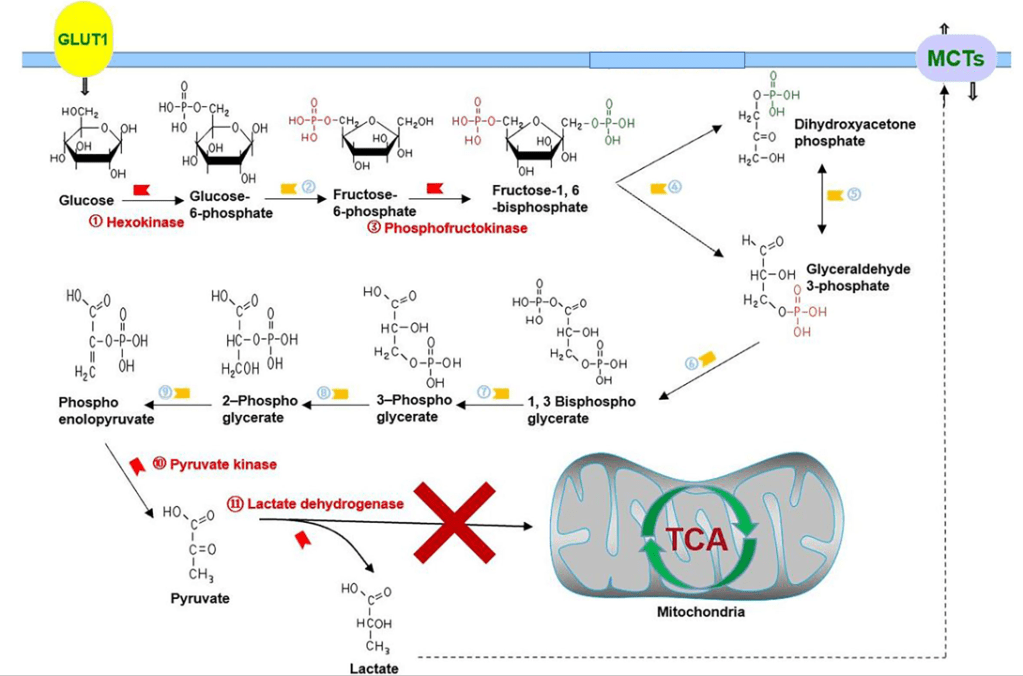
La glycolyse est nécessaire non seulement pour produire de l’ATP, mais aussi pour générer des intermédiaires métaboliques nécessaires à la production d’éléments essentiels à la prolifération cellulaire, tels que les acides aminés, les lipides et les nucléotides.
Alors que les cellules différenciées expriment l’isoforme M 1 de la pyruvate kinase (PK-M 1), des cellules cancéreuses expriment l’isoforme M2, qui est normalement exprimée uniquement au cours du développement embryonnaire. PK-M2 est activée par la signalisation de la tyrosine kinase et entraîne la conversion du pyruvate en lactate plutôt que d’orienter le pyruvate vers le cycle de Krebs.
La chémorésistance des tumeurs semble associée à une augmentation de l’effet Warburg. Par exemple, la résistance aux traitements 5-FU/cisplatine dans le cancer colorectal est associée à une augmentation de la glycolyse par augmentation du ratio PK-M2/PK-M1. Cet effet est médié par les lncARN Xist/miR-137 (Hailun et al., 2021).
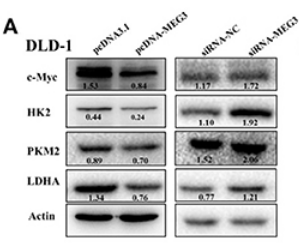
La lactate déshydrogénase A qui catalyse la transformation du pyruvate en lactate a son expression fréquemment augmentée dans les cellules tumorales. Le gène codant cette enzyme est une cible directe de l’oncogène et facteur de transcription c-Myc (Shim et al. 1997). L’inhibition de l’expression de la lactate déshydrogénase A par des ARN interférents inhibe la tumorigenèse (Fantin et al., 2006). Le long ARN non codant MEG3 réduit la stabilité de la protéine c-Myc en stimulant son ubiquitinylation et sa dégradation. Cela a pour conséquence de diminuer la prolifération des cellules tumorales et notamment en inhibant la glycolyse et la production de lactate (Zuo et al., 2020). L’expression de MEG3 est diminuée dans de nombreux cas de cancers colo-rectaux et il peut être considéré comme un suppresseur de tumeur.
Il existe des mécanismes inhibiteurs de l’effet Warburg. Par exemple, le long ARN non codant (lncARN) MEG3 dont l’expression est diminuée dans les cellules des tumeurs colo-rectales est capable d’inhiber la glycolyse en activant (indirectement) l’ubiquitinylation et la dégradation de c-Myc qui est nécessaire à l’expression d’enzymes telles que PK-M2, hexokinase 2 et la lactate déshydrogénase A (Zuo et al., 2020). L’expression de MEG3 est activée par la vitamine D ce qui pourrait avoir des applications thérapeutiques intéressantes.
Signalons que l’effet Warburg n’est pas une propriété générale et que des cellules tumorales ont été caractérisées avec toujours une dépendance majeure aux phosphorylations oxydatives dans les mitochondries (Pendleton et al., 2023). Il peut exister une hétérogénéité au sein d’une même tumeur avec des cellules avec des cellules à profil « glycolytique » et des cellules à profil dit OxPhos (phosphorylation oxydative dans la mitochondrie) (Kim et al., 2019). Il a même été montré que des cellules tumorales en déficit de mitochondries peuvent récupérer ces organites par transfert à partir des cellules stromales voisines (Novak et al., 2025).
Le métabolisme lipidique est aussi altéré dans les cellules tumorales. Le métabolisme des cellules normales quiescentes dépend généralement de l’absorption des lipides de la circulation. Les cellules tumorales très prolifératives montrent une forte avidité à acquérir des lipides et un taux de cholestérol élevés, en augmentant l’absorption de lipides et de lipoprotéines exogènes (ou alimentaires), mais aussi en activant leurs mécanismes de synthèse lipidique endogène, notamment la lipogenèse de novo à partir de sources non lipidiques telles que le glucose et l’acétate (Bian et al., 2020). Cette production accrue dans les cellules tumorales fournit des lipides pour la synthèse des membranes et des molécules de signalisation lors de la prolifération cellulaire rapide et de la croissance, dans un contexte où la disponibilité des lipides est limitée dans le microenvironnement tumoral. La synthèse lipidique est contrôlée par plusieurs facteurs de transcription, tels que SREBP1 et SREBP2 (SREBP1/2) (Griffith et al., 2013; Jiang et al., 2020).
Les facteurs oncogènes, tels KRAS et PI3K, favorisent la lipogenèse de novo dans le cancer du sein et d’autres types de cancer par l’activation de mTORC1. La protéine Lipine-1 séquestre SREBP1/2 mature dans une région du noyau séparé de l’ADN, empêchant ainsi SREBP1/2 d’activer l’expression des gènes. mTORC1 phosphoryle directement la Lipine-1, ce qui inhibe sa translocation nucléaire et restaure ainsi l’activité de SREBP1/2 (Peterson et al., 2011).
La capacité à faire former de nouveaux vaisseaux sanguins
La progression tumorale nécessite la mise en place de nouveaux vaisseaux ce qui permet d’apporter du glucose et de l’O2 à la nouvelle masse de cellules en prolifération active. Ce phénomène est appelé angiogenèse tumorale. Il passe par la production de facteurs angiogéniques qui stimulent les cellules endothéliales des vaisseaux sanguins préexistants à s’engager dans une angiogenèse (Folkman, 2007). Par exemple, des stimulateurs angiogéniques tels que le VEGF sont libérés par les cellules tumorales, et parfois par les macrophages du microenvironnement tumoral. L’angiogenèse est également associée à la propagation des tumeurs aux sites métastatiques.
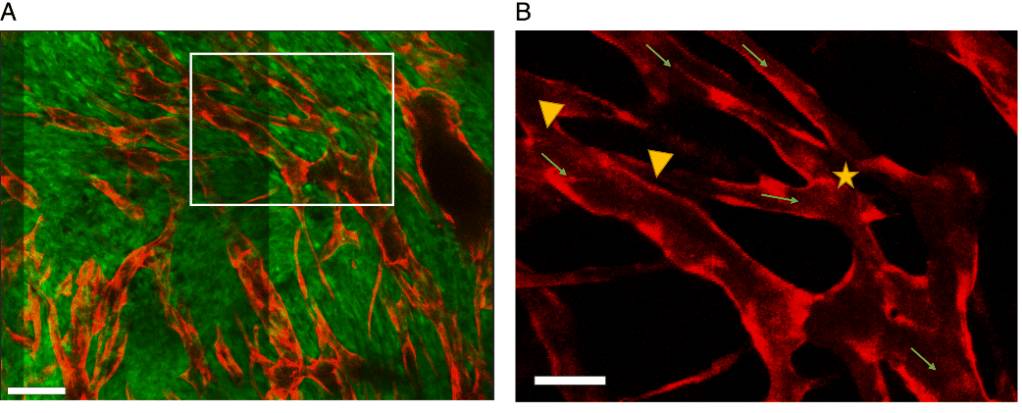
Les vaisseaux tumoraux sont caractérisés par une morphologie anormale : ils sont trop branchés et aussi perméables, car ils sont dépourvues de péricytes et de cellules musculaires lisses et possèdent une membrane basale irrégulière avec un revêtement endothélial irrégulier. Ils sont ainsi fortement dysfonctionnels dans leurs propriétés de barrière et de transport.
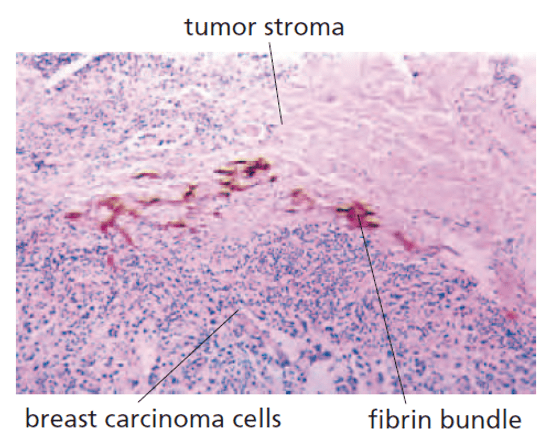
Cela aboutit à une hétérogénéité d’oxygénation dans les tumeurs qui contribue à l’hétérogénéité phénotypique des cellules tumorales (Barnabeu et al., 2020).
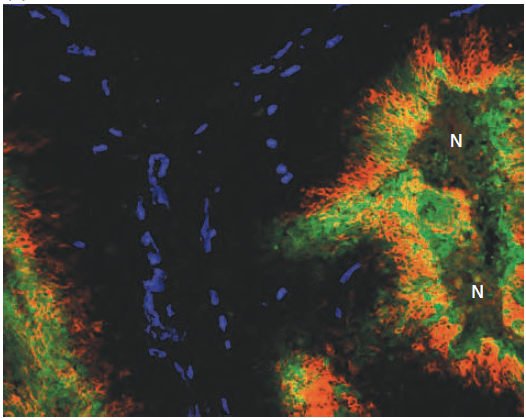
là où les vaisseaux sanguins (taches bleues) assurent une bonne oxygénation, le
la tumeur apparaît noire. En revanche, dans les zones de mauvaise vascularisation et avec une hypoxie modérée, une enzyme anhydrase carbonique est exprimée (révélée en rouge orange), tandis que dans les zones d’hypoxie importante, le colorant pimonidazole est détecté (vert). Le chevauchement entre ces deux marqueurs apparaît en orange. Des zones de nécrose, situées encore plus loin du système vasculaire tumoral, sont indiqués par « N ». Source : https://aacrjournals.org/cancerres/article/62/23/7066/509392/Pimonidazole-Binding-and-Tumor-Vascularity-Predict
Des stratégies thérapeutiques visant à induire un changement phénotypique dans les vaisseaux tumoraux pour qu’ils ressemblent à des vaisseaux normaux, appelées normalisation vasculaire, ont été testées (Huang et al., 2013). Les anticorps anti-VEGF induisent une normalisation vasculaire dans les modèles précliniques et en clinique, ce qui peut expliquer en partie leur efficacité dans le traitement des cancers métastatiques. Après traitement anti-VEGF, les vaisseaux tumoraux montrent aussi une augmentation de la perfusion et de l’efficacité des chimiothérapies antitumorales (Goel et al., 2011).
Cependant, de manière un peu paradoxale, les régions tumorales bien oxygénées répondent mieux à la radiothérapie (Bertout et al., 2011). L’hypoxie provoque en effet des modifications dans l’expression des gènes et la sélection clonale associée aboutit à des tumeurs aux phénotypes plus agressifs et métastatiques. C’est pour cela que certains traitements anti-angiogéniques peuvent parfois aboutir à de mauvais résultats. L’un des responsables est le facteur de transcription HIF-1. C’est un régulateur clé qui intervient dans les réponses adaptatives aux changements d’oxygénation par l’activation de la transcription de gènes impliqués dans le métabolisme du glucose et la survie cellulaire. HIF-1 se compose de deux sous-unités (α et β) et HIF-1α est hydroxylé et dégradé dans des conditions de normoxie, mais s’accumule dans des conditions d’hypoxie ce qui permet d’activer HIF-1. L’existence d’inhibiteur de HIF-1 comme TiPARP (qui réalise des ADP-ribosylation, une modification post-traductionnelle) offre des espoirs d’améliorer les thérapies, y compris en conditions hypoxiques (Zhang et al., 2020).
La capacité de bloquer les processus immunitaires anti-tumoraux
Comme le montre l’expérience qui suit, le système immunitaire a la capacité de s’attaquer à des cellules tumorales :
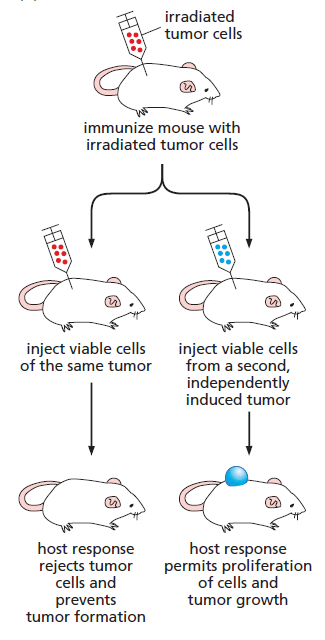
La capacité du système immunitaire à reconnaître et à éliminer les cellules cancéreuses est un déterminant essentiel de la croissance tumorale et du pronostic vital pour les patients.
Cependant, beaucoup de tumeurs échappent activement au système immunitaire en induisant un état immunosuppresseur (Zitvogel et al., 2006). Les lymphocytes infiltrant les tumeurs CD8 (TIL) montrent souvent des fonctions effectrices diminuées. PD-1 et CTLA-4 sont deux récepteurs inhibiteurs des fonctions effectrices de ces lymphocytes qui jouent normalement un rôle dans l’immunotolérance. Ils sont fréquemment ciblés par les cellules tumorales pour atténuer les fonctions effectrices via leurs ligands, PDL-1 et B7 (Topalian et al., 2015). Une plus forte expression de PDL-1 dans les cellules tumorales est associée à un mauvais pronostic (Okazaki et al., 2007). Des immunothérapies anti-PD1 ont permis de remporter certains succès notamment pour la lutte contre les mélanomes et les tumeurs pulmonaires (Iwai et al., 2017).
Le système immunitaire peut paradoxalement être impliqué dans des environnements protumorigéniques :
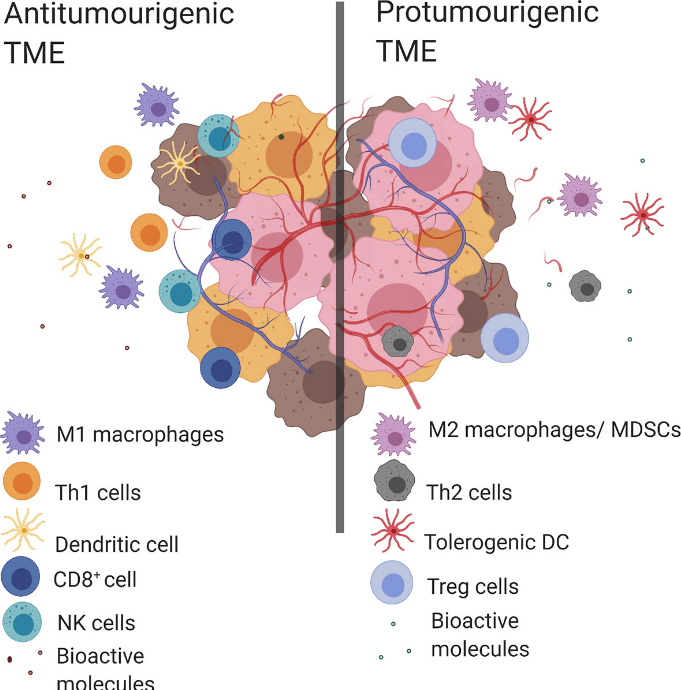
Des macrophages s’accumulent aussi au cours de la progression tumorale proviennent de l’extravasation de monocytes dérivés de la moelle osseuse (DeNardo et al. 2019). Les macrophages peuvent acquérir un spectre de phénotypes pro-inflammatoires (également appelés « M1 ») à anti-inflammatoires (également appelés « M2 ») (Mantovani et al., 2017). Dans les tumeurs, les macrophages pro-inflammatoires freinent initialement la croissance tumorale en activant les lymphocytes cytotoxiques. Cependant, au cours de la progression tumorale, les macrophages sont biaisés vers un phénotype anti-inflammatoire favorisant la malignité tumorale en supprimant les lymphocytes cytotoxiques et en stimulant la formation de vaisseaux sanguins (Squadrito et al., 2012).
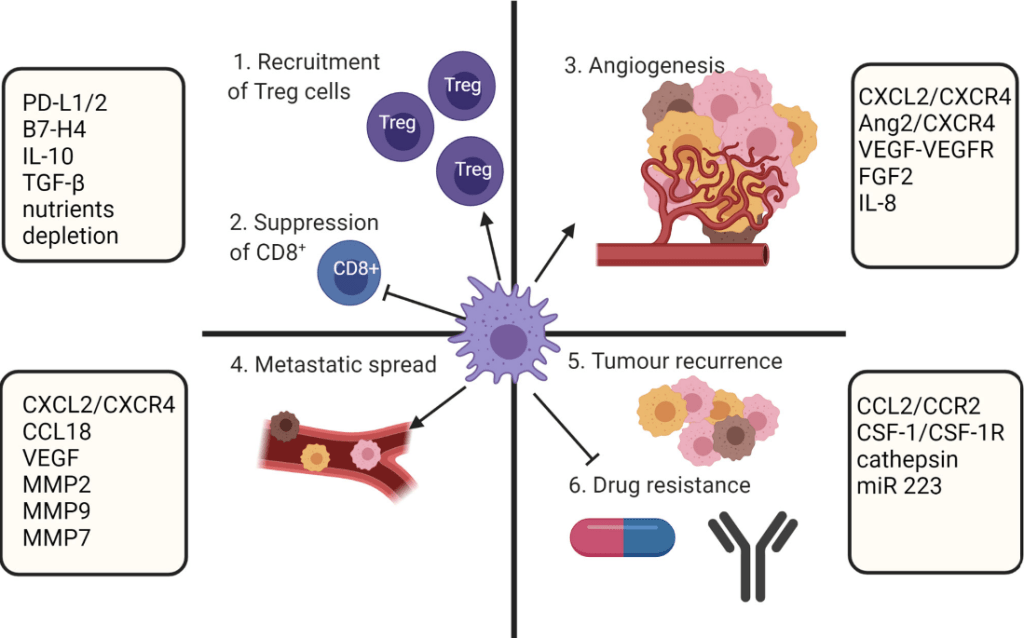
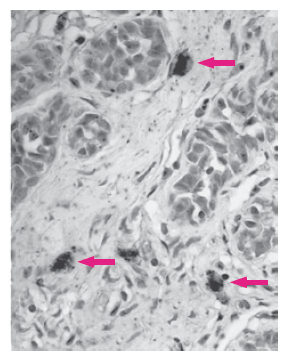
Ces macrophages de type M2 sont caractérisés par une forte expression du récepteur au mannose MRC1/CD206 et par une faible expression de l’intégrine αX (CD11c). L’accumulation de ces macrophages largement anti-inflammatoires est donc corrélée à un mauvais pronostic dans la plupart des cancers (Mahmoud et al., 2012). Les mécanismes qui amènent les macrophages à devenir de type M2 commencent à être élucidées (Hung et al., 2023).
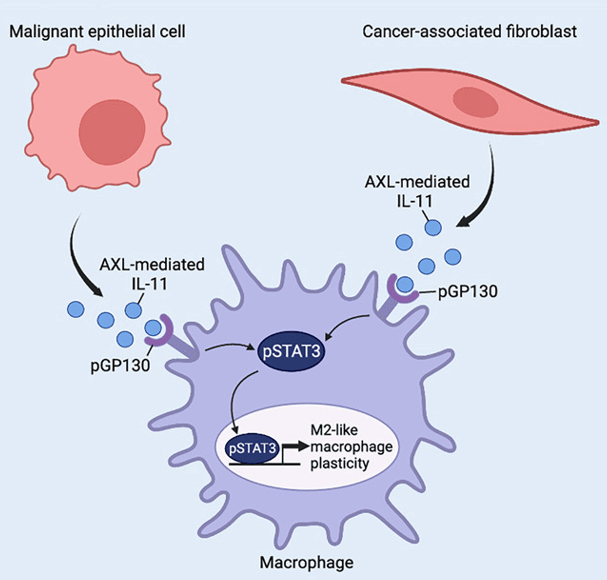
On voit donc qu’en plus d’un blocage des mécanismes immunitaires pouvant les attaquer, les tumeurs détournent à leur profit des processus liés à l’immunité (comme l’angiogenèse).
EN DIRECT DES LABOS :
QUELQUES EQUIPES FRANCOPHONES QUI TRAVAILLENT SUR LE SUJET :
Remodelage de la chromatine, réparation de l’ADN et épigénétique – Institut Gustave Roussy, Villejuif
Immunologie intégrative des tumeurs et immunothérapie du cancer – Institut Gustave Roussy, Villejuif
- Adhérences cellule-cellule
- Arabidopsis thaliana
- Axe antéro-postérieur chez la drosophile
- Biomécanique du développement
- Caenorhabditis elegans
- Concepts principaux
- Contrôle de la traduction
- Contrôle de la transcription
- Contrôle génétique et épigénétique
- Croissance du tube pollinique et double fécondation chez les Angiospermes
- Croissance et guidage axonal
- Des modèles animaux moins classiques
- Développement de l’oeil des Vertébrés
- Développement et évolution
- Et l’Humain ?
- Exercices sur l’ovogenèse, la spermatogenèse et la fécondation
- Exercices sur le contrôle de l’expression des gènes
- Exercices sur le développement des bourgeons de membre
- Exercices sur le développement des muscles striés squelettiques
- Exercices sur le développement des végétaux et les hormones végétales
- Exercices sur les cycles et les divisions cellulaires
- Exercices sur les étapes du développement, les inductions embryonnaires et la mise en place des axes de polarité
- Exercices sur les matrices extracellulaires, le cytosquelette et les adhérences cellule-cellule
- Exercices sur les voies de signalisation
- Glossaire
- Glossaire des termes liés à la génétique
- Glossaire des termes liés au cytosquelette, la matrice extracellulaire, l’adhérence et la migration cellulaire
- Hématopoïèse et développement des cellules du système immunitaire
- Histoire de la biologie cellulaire et de la biologie du développement
- L’acide rétinoïque
- L’apoptose
- L’autophagie
- L’organogenèse
- L’ovogénèse prépare le développement embryonnaire
- La drosophile
- La fécondation
- La formation des somites
- La gastrulation
- La gastrulation (version allégée)
- La métamorphose chez les Hexapodes et les Amphibiens
- La neurogénèse chez les mammifères adultes
- La neurulation
- La poule
- La signalisation calcique
- La souris
- La superfamille TGFβ et ses voies de signalisation
- La voie de signalisation de l’auxine et ses rôles
- La voie de signalisation Hedgehog
- La voie de signalisation Hippo et ses composants YAP/TAZ
- La voie de signalisation Notch
- Le clivage
- Le cytosquelette
- Le destin des cellules et les réseaux de régulation génique
- Le développement des bourgeons de membre
- Le développement des muscles striés squelettiques
- Le développement des organes génitaux et des cellules germinales
- Le développement du cortex
- Le méristème apical caulinaire en phase végétative et lors de la formation d’une fleur
- Le poisson zèbre
- Le xénope
- Les cellules des crêtes neurales
- Les cellules et les gènes en action dans le développement
- Les cellules souches
- Les cycles et les divisions cellulaires
- Les étapes du développement
- Les étapes du développement embryonnaire d’Arabidopsis thaliana et leur contrôle
- Les inductions embryonnaires et les gradients de morphogène
- Les matrices extracellulaires animales
- Les organismes modèles
- Les outils pour étudier l’expression et la fonction des gènes
- Les parois des cellules végétales
- Les techniques et les outils pour la biologie cellulaire
- Les transitions épithélio-mésenchymateuses et les migrations cellulaires
- Les vésicules extracellulaires
- Les voies de signalisation
- Les voies de signalisation FGF
- Mise en place des axes chez les Vertébrés
- Structures et processus cellulaires
- Voies de signalisation WNT

{kind=link}
{kind=link}