Par Patrick Pla, Université Paris-Saclay
L’autophagie représente une voie de dégradation de composants cellulaires par les lysosomes (ou les vacuoles chez les cellules végétales). C’est un processus physiologique qui permet de se débarrasser de composants obsolètes ou défectueux et permet de recycler des molécules utiles. Elle est notamment déclenchée en période de pénurie de nourriture.

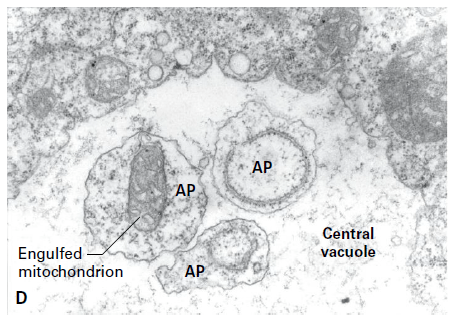
L’autophagie a été observée pour la première fois grâce aux progrès de la microscopie électronique dans les années 1950 et 1960 mais ce n’est qu’à partir des années 1990 que sa fonction et son mécanisme ont été compris. De l’autophagie a été décelée dans de très nombreuses circonstances. Par exemple, au cours de la fécondation chez les animaux, quelques mitochondries paternelles entrent dans le cytoplasme de l’ovocyte. Ces mitochondries sont éliminées par autophagie, ce qui assure l’exclusivité de l’origine maternelle des mitochondries d’un embryon (Sato et al., 2011). Autre cas, lorsque des cellules subissent des dommages à l’ADN conséquents, des fragments de chromosome sont expulsés du noyau et forment des micronoyaux. Ces micronoyaux sont éliminés par autophagie (dans ce cas, elle est appelée nucléophagie) (Mucino-Hernandez et al., 2023).
L’autophagie a également été observée comme un élément crucial du maintien des cellules souches et comme s’opposant au vieillissement chez l’hydre (Cnidaire) (Tomczyk et al., 2020).
Chez les végétaux, les plantes privées d’autophagie (mutants atg) sont très sensibles au manque de carbone et d’azote et présentent une sénescence précoce et une croissance réduite (Doelling et al. 2002; Li et al. 2015; Barros et al. 2017), ainsi que des retards dans la germination (Yoshimoto et al. 2014; Avin-Wittenberg et al. 2015).
Les protéines clés contrôlant l’autophagie sont codés par les Atg (pour autophagy-related genes), initialement découverts chez la levure, mais très conservés au cours de l’évolution. En inhibant l’expression des gènes Atg, on a pu étudier l’impact de l’autophagie sur diverses fonctions physiologiques de divers modèles cellulaires et d’organisme : l’adaptation à la carence en nutriments, le renouvellement des constituants cellulaires, la limitation du vieillissement, l’homéostasie des organites et la réponse immunitaire, ainsi que la relation entre l’autophagie et les maladies humaines telles que les maladies neurodégénératives, le diabète, le cancer et les infections.
Les matériaux cellulaires à dégrader par l’autophagie sont séquestrés dans des vésicules à double membrane appelées « autophagosomes » et transportés vers les lysosomes des cellules de mammifères ou les vacuoles chez les levures et les végétaux. Le matériel membranaire formant les autophagosomes provient de divers sources : membrane plasmique le plus souvent mais aussi endosomes de recyclage, appareil de Golgi, réticulum endoplasmique et même membrane externe mitochondriale (Ravikumar et al., 2010; Hailey et al., 2010). Ainsi, la production d’autophagosomes est liée à la dynamique des compartiments endomembranaires et notamment de l’endocytose dépendante de la clathrine.
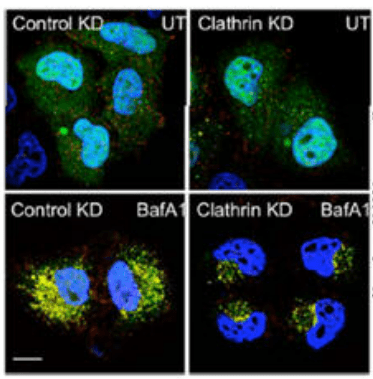
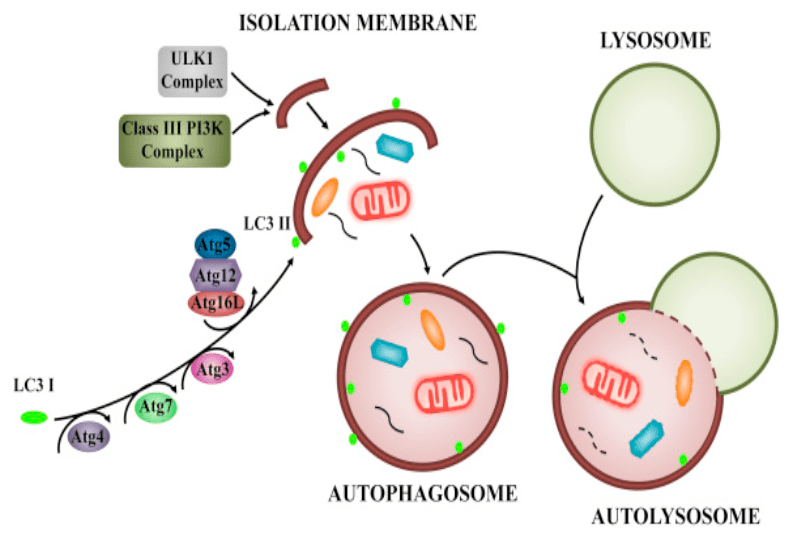
Pour initier la biogenèse des autophagosomes, plusieurs complexes ULK sont assemblés et l’ensemble fonctionne comme un échafaudage pour le recrutement d’autres protéines ATG. Le complexe ULK/ATG permet le recrutement et l’activation de complexes PI3K (phosphatidylinositol 3-kinase) qui permettent l’incorporation du phosphatidylinositol 3‑phosphate (PI3P, à ne pas confondre avec le le phosphatidylinositol 3,4,5‑trisphosphate (PIP3)) dans les membranes à partir desquelles les autophagosomes sont générés. Ensuite, les protéines WIPI 1-4 se lient au PI3P et recrutent les deux prochains composants importants de l’expansion des phagophores : les protéines ATG2 et le complexe ATG16L1 (Allmanai et al., 2022).
Il existe deux principaux types d’autophagie, qui diffèrent par le mode de séquestration des cibles à dégrader. Dans l’autophagie non sélective, les composants cytoplasmiques qui se trouvent sur les sites de formation des autophagosomes sont séquestrés de manière aléatoire. En revanche, dans l’autophagie sélective, certaines molécules, structures et organites sont reconnus par des protéines spécifiques appelées « récepteurs de l’autophagie » et sont sélectivement engloutis par les autophagosomes.
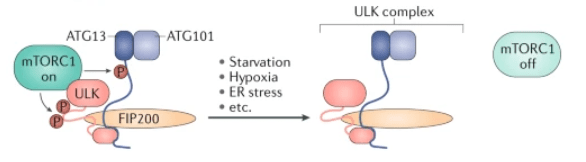
La biogenèse des autophagosomes est induite par une grande variété de signaux provenant de l’intérieur et de l’extérieur des cellules, notamment le manque de nutriments et d’ions (acides aminés, glucose, fer, zinc, phosphate, etc.), différents types de stress (stress du réticulum endoplasmique (RE), stress oxydatif, stress hypoxique, dommages à l’ADN, etc.), la formation d’agrégats protéiques anormaux ou d’organites endommagés et l’infection microbienne. Ces signaux sont transmis via différentes voies de signalisation, mais beaucoup d’entre eux convergent vers l’inactivation du complexe TORC1.
Tant que TORC1 est actif, l’initiation de l’autophagie est inhibée par sa phosphorylation des protéines ULK et ATG13. Lorsque TORC1 n’est pas activé (dans les conditions vues au paragraphe précédent), ULK et ATG13 ne sont plus phosphorylés et l’assemblage de la membrane d’isolement peut commencer.
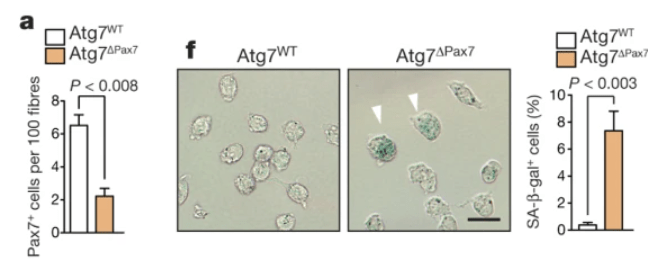
Des études dans les cellules souches musculaires adultes (les cellules satellites) ont montré que l’inhibition de TORC1 par la rapamycine peut activer l’autophagie et éviter la sénescence liée à l’âge dans ces cellules souches (Garcia-Prat et al., 2016). Si on élimine l’expression d’Atg7 dans ces cellules et donc on inhibe l’autophagie, il y a une augmentation de la sénescence et une chute importante du nombre de cellules satellites par fibre musculaire.
Une fois l’autophagosome formé, il doit fusionner avec les lysosomes. Les protéines GABARAP recrutent la forme palmitoylée de PI4KIIα, une lipide kinase qui génère le phosphatidylinositol 4-phosphate (PI4P). L’inhibition de GABARAP ou de PI4KIIα, ou la surexpression d’un mutant PI4KIIα dominant négatif, diminue le flux d’autophagie en bloquant la fusion autophasome:lysosome, ce qui entraîne l’accumulation d’autophagosomes anormalement grands (Wang et al., 2015).
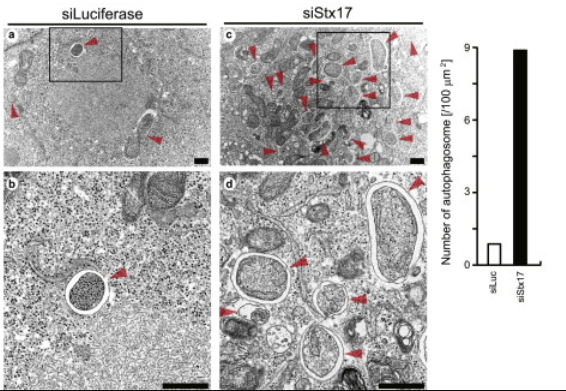
Les protéines SNARE sont également essentielles à la fusion autophagosome:lysosome. Les protéines SNARE sont des protéines membranaires qui se lient à la membrane soit par leur domaine transmembranaire situé à l’extrémité C-terminale, soit par des chaînes carbonées hydrophobes associées à des résidus cystéine. Les protéines SNARE sont classées selon une séquence au centre de leur domaine SNARE en R-SNARE et Q-SNARE (Fasshauer et al., 1998; Hong, 2005). Pendant la fusion, les R- et Q-SNARE des membranes en vis-à-vis forment un faisceau de quatre hélices α. L’énergie libérée lors du regroupement de ces complexes rapproche les membranes et permet le mélange lipidique et la fusion des membranes (Li et al., 2007). Plus précisément, dans le processus de fusion membranaire autophagosome-lysosome chez les mammifères, le premier ensemble de SNARE identifiés est constitué de STX17 (la syntaxine-17 de type Q-SNARE) sur la membrane autophagosomale, de SNAP29 (aussi de type Q-SNARE) et de VAMP8 (R-SNARE) sur la membrane lysosomale (Ikatura et al., 2012).
Le processus de fusion est également soutenu par les GTPases de type Rab (D’Agostino et al., 2017).
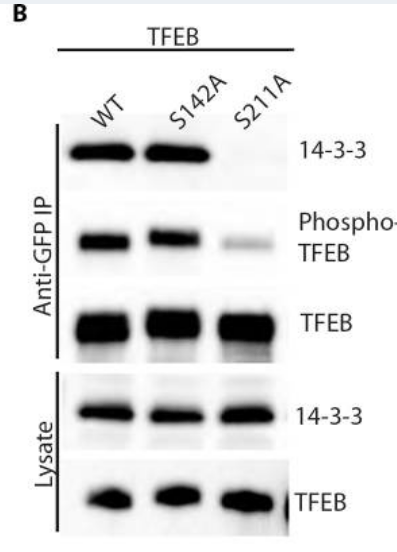
Très logiquement, la genèse de lysosomes et d’autophagosomes est souvent coordonnée. Le facteur de transcription à hélice-boucle-hélice/leucine zipper TFEB est un régulateur principal de la biogenèse des lysosomes et de l’autophagie. En conditions normales, TFEB subit une phosphorylation médiée par mTOR et reste dans le cytosol. L’action de facteurs de croissance ou la présence d’une quantité suffisante d’acides aminés active en effet mTOR qui inhibe l’autophagie en phosphorylant TFEB. Lorsqu’il y a pénurie de nutriments et que mTOR n’est plus activé ou lorsque les conditions de stockage dans les lysosomes deviennent saturées, TFEB est déphosphorylé puis est transloqué dans le noyau où il active ses gènes cibles via le système CLEAR (pour Coordinated Lysosomal Expression and Regulation). Contrairement aux activateurs purement autophagiques, TFEB favorise la clairance en régulant plusieurs étapes de la chaine de dégradation lysosomale-autophagique, notamment la formation de lysosome, la formation d’autophagosomes et la fusion autophagosome-lysosome (Sardiello et al., 2009, Song et al., 2021).

La rapamycine qui inhibe mTOR stimule l’autophagie, ce qui contribue à éliminer les agrégats de protéines intracellulaires qui s’accumulent dans de nombreuses maladies neurodégénératives et à réduire leur toxicité (Menzies et al., 2015).
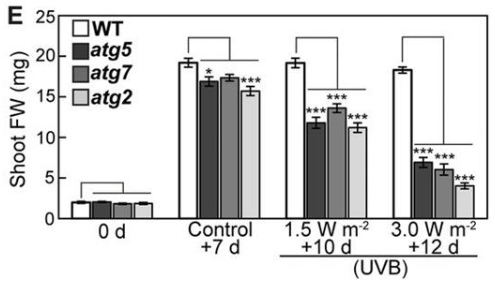
Des perturbations de l’autophagie participent aux mécanismes du vieillissement (Aman et al., 2021). Une grande partie des cellules souches hématopoïétiques (HSC) âgées présentent des niveaux d’autophagie diminués. Cela entraîne l’accumulation de mitochondries, ce qui à son tour induit un stress métabolique. Des niveaux élevés d’espèces réactives de l’oxygène, générées par les mitochondries, s’accumulent dans les HSC et compromettent leur fonctionnement (Ito et al., 2006, Mohrin et al., 2016). Chez les végétaux comme Arabidopsis thaliana, des mutants avec une perte-de-fonction de l’autophagie ont des feuilles qui subissent un vieillissement acceléré. La dégradation autophagique des chloroplastes, appelée chlorophagie, élimine les chloroplastes âbimés par les UVB ou une forte intensité de lumière dans le visible (Izumi et al. 2017, Nakamura et al. 2018).
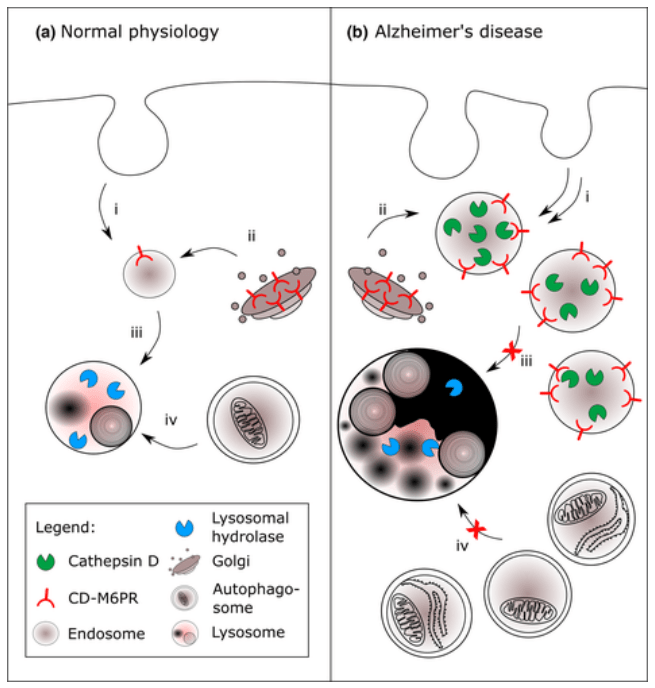
Des perturbations de l’autophagie sont observées dans de nombreuses pathologies. Par exemple dans la maladie d’Alzheimer, les autophagosomes n’arrivent pas à fusionner avec les lysosomes ce qui crée un engorgement (Whyte et al., 2016). Ces dysfonctionnements peuvent précéder de plusieurs années l’apparition des symptomes de la maladie d’Alzheimer (Cataldo et al., 2000).
EN DIRECT DES LABOS :
- Adhérences cellule-cellule
- Arabidopsis thaliana
- Axe antéro-postérieur chez la drosophile
- Biomécanique du développement
- Caenorhabditis elegans
- Concepts principaux
- Contrôle de la traduction
- Contrôle de la transcription
- Contrôle génétique et épigénétique
- Croissance du tube pollinique et double fécondation chez les Angiospermes
- Croissance et guidage axonal
- Des modèles animaux moins classiques
- Développement de l’oeil des Vertébrés
- Développement et évolution
- Et l’Humain ?
- Exercices sur l’ovogenèse, la spermatogenèse et la fécondation
- Exercices sur le contrôle de l’expression des gènes
- Exercices sur le développement des bourgeons de membre
- Exercices sur le développement des muscles striés squelettiques
- Exercices sur le développement des végétaux et les hormones végétales
- Exercices sur les cycles et les divisions cellulaires
- Exercices sur les étapes du développement, les inductions embryonnaires et la mise en place des axes de polarité
- Exercices sur les matrices extracellulaires, le cytosquelette et les adhérences cellule-cellule
- Exercices sur les voies de signalisation
- Glossaire
- Glossaire des termes liés à la génétique
- Glossaire des termes liés au cytosquelette, la matrice extracellulaire, l’adhérence et la migration cellulaire
- Hématopoïèse et développement des cellules du système immunitaire
- Histoire de la biologie cellulaire et de la biologie du développement
- L’acide rétinoïque
- L’apoptose
- L’organogenèse
- L’ovogénèse prépare le développement embryonnaire
- La drosophile
- La fécondation
- La formation des somites
- La gastrulation
- La gastrulation (version allégée)
- La métamorphose chez les Hexapodes et les Amphibiens
- La neurogénèse chez les mammifères adultes
- La neurulation
- La poule
- La signalisation calcique
- La souris
- La superfamille TGFβ et ses voies de signalisation
- La voie de signalisation de l’auxine et ses rôles
- La voie de signalisation Hedgehog
- La voie de signalisation Hippo et ses composants YAP/TAZ
- La voie de signalisation Notch
- Le clivage
- Le cytosquelette
- Le destin des cellules et les réseaux de régulation génique
- Le développement des bourgeons de membre
- Le développement des muscles striés squelettiques
- Le développement des organes génitaux et des cellules germinales
- Le développement du cortex
- Le méristème apical caulinaire en phase végétative et lors de la formation d’une fleur
- Le poisson zèbre
- Le xénope
- Les cellules des crêtes neurales
- Les cellules et les gènes en action dans le développement
- Les cellules souches
- Les cellules tumorales
- Les cycles et les divisions cellulaires
- Les étapes du développement
- Les étapes du développement embryonnaire d’Arabidopsis thaliana et leur contrôle
- Les inductions embryonnaires et les gradients de morphogène
- Les matrices extracellulaires animales
- Les organismes modèles
- Les outils pour étudier l’expression et la fonction des gènes
- Les parois des cellules végétales
- Les techniques et les outils pour la biologie cellulaire
- Les transitions épithélio-mésenchymateuses et les migrations cellulaires
- Les vésicules extracellulaires
- Les voies de signalisation
- Les voies de signalisation FGF
- Mise en place des axes chez les Vertébrés
- Structures et processus cellulaires
- Voies de signalisation WNT
